
Abstract
The C-C motif chemokine receptor 1 (CCR1) plays key roles in guiding leukocyte movement during immune surveillance and inflammatory responses. Targeting CCR1 is a promising approach for treating autoimmune diseases and cancers. We previously developed monoclonal antibodies against mouse CCR1 (mCCR1) (clones C1Mab-2 and C1Mab-6) for use in flow cytometry and Western blotting. However, the specific binding sites have not yet been identified. This study examined the binding epitope of C1Mab-2 and C1Mab-6 using flow cytometry. Analysis of mCCR1 mutants with altered extracellular domains showed that C1Mab-2 and C1Mab-6 bind to the N-terminal region of mCCR1. Additionally, PA-tag substitution experiments identified the epitope as 1st Met and amino acids 2–13 of mCCR1. Further alanine (or glycine) scanning within the N-terminal region (amino acids 2–13) demonstrated that Glu2, Asp5, and Phe6 are essential for recognition by C1Mab-2, and Glu2, Ile3, and Asp5 are crucial for recognition by C1Mab-6 in flow cytometry and Western blotting. These findings contribute to the understanding of mCCR1 and C1Mab interaction.
Introduction
Chemokine receptors are part of the seven-transmembrane G protein–coupled receptor family and play essential roles in controlling leukocyte trafficking during immune surveillance and inflammation responses. 1 Chemokines are classified into the CC, CXC, XC, and CX3C subfamilies based on the arrangement of the first two conserved cysteine residues. CC chemokines (CCL1–CCL28) are identified by C-C chemokine receptors CCR1–CCR10. 2 Upon ligand engagement, chemokine receptors typically activate heterotrimeric G protein signaling pathways and recruit β-arrestins for receptor regulation endocytosis.3,4
CCR1 is a crucial mediator of inflammatory responses and plays a role in the development of autoimmune diseases.1,5 Accordingly, CCR1 is considered a promising therapeutic target for allergic and autoimmune disorders. 6 Notably, CCR1 shows ligand promiscuity, allowing it to recognize multiple CC chemokines, including CCL3, CCL5, CCL7, CCL8, CCL13–16, and CCL23.2,7,8
Structural elucidation of chemokine receptor activation is essential for rational drug design targeting the chemokine system. Among CCR family members, both inactive and ligand-bound active conformations have been resolved for CCR2 and CCR5.9–12 Additionally, ligand-bound active-state structures of CCR6 and CCR8, along with inactive-state structures of CCR7 and CCR9, have been reported.13–16 The structure of the CCL15–CCR1 complex showed that extracellular loops (ECL2 and ECL3) contain key factors for ligand recognition, setting CCR1 apart from other chemokine receptor–ligand interactions and offering new insights into chemokine-binding mechanisms. 9 Furthermore, structural analyses of CCR8 in complex with either its endogenous ligand CCL1 or an antagonistic monoclonal antibody (mAb) have elucidated mechanisms underlying receptor activation and antibody-mediated inhibition. 13 These findings highlight the usefulness of antichemokine receptor mAbs with specific epitopes for structural and functional studies.
The Cell-Based Immunization and Screening (CBIS) method is a strategy for generating a variety of mAbs against membrane proteins. In this approach, antigen-overexpressed cells are immunized, and the hybridoma supernatants are then screened using flow cytometry-based high-throughput techniques. We have developed several mAbs targeting chemokine receptors, such as mouse CCR1 (mCCR1) and mouse CCR5 (mCCR5), utilizing the CBIS method.17,18 These mAbs are expected to recognize various epitopes, including linear and conformational ones. However, their specific binding epitopes are still undefined.
We previously reported the flow cytometry-based epitope mapping of an anti-mCCR1 mAb (S15040E) and an anti-mCCR8 mAb (C8Mab-2) using mutants with the extracellular domain substituted and alanine scanning.19,20 Using the epitope identification strategy, we aimed to determine the binding epitopes of anti-mCCR1 mAbs (C1Mab-2 and C1Mab-6) through flow cytometry and Western blotting.
Materials and Methods
Plasmid construction
pCAG-Ble-mCCR1 and pCAG-Ble-mouse CCR5 (mCCR5) were generated as previously described.17,18 Chimeric mutants, including mCCR5 (mCCR1p2–34), mCCR5 (mCCR1p92–107), mCCR5 (mCCR1p172–197), and mCCR5 (mCCR1p265–281), were produced as described previously. 19 Alanine (or glycine)-substituted mutants of mCCR1 were generated using QuikChange Lightning Site-Directed Mutagenesis Kits (Agilent Technologies Inc., Santa Clara, CA, USA). PA-tag (GVAMPGAEDDVV)-substituted mCCR1 mutants were created using a PCR-based method. The PCR fragments containing the desired mutations were inserted into the pCAG-Ble or pCAG-neo vectors (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan).
Cell line and plasmid transfection
The chimeric, PA-tag-substituted, and point-mutant plasmids were transfected into Chinese hamster ovary (CHO)-K1 cells (American Type Culture Collection, Manassas, VA, USA) using the Neon Transfection System (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Antibodies
C1Mab-2 and C1Mab-6 were established as described previously. 18 C5Mab-2 and C5Mab-4 were also developed by our laboratory.17,21 An anti-mCCR1 mAb (clone S15040E) was purchased from BioLegend (San Diego, CA, USA).
Flow cytometry
Cells were collected after brief exposure to 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (Nacalai Tesque, Inc.). After washing with 0.1% bovine serum albumin in phosphate-buffered saline, 2 × 105 cells were treated with 1 µg/mL of C1Mab-2, C1Mab-6, C5Mab-2, C5Mab-4, or S15040E for 30 minutes at 4°C, followed by incubation with Alexa Fluor 488-conjugated anti-rat IgG (1:2,000; Cell Signaling Technology, Inc., Danvers, MA, USA). Fluorescence data (from a total of 5000 cells per sample) were collected using the SA3800 Cell Analyzer (Sony Corp., Tokyo, Japan). Using FlowJo software (BD Biosciences, Franklin Lakes, NJ, USA), single cells were gated based on side scatter versus forward scatter, and the fluorescence intensity was plotted.
Western blotting
Cell lysates were boiled in sodium dodecyl sulfate (SDS) sample buffer (Nacalai Tesque, Inc.). Proteins (10 µg/lane) were electrophoresed on 5%–20% polyacrylamide gels (FUJIFILM Wako Pure Chemical Corporation) and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck KGaA, Darmstadt, Germany). After blocking with 4% nonfat milk (Nacalai Tesque, Inc., Kyoto, Japan), PVDF membranes were incubated with 1 µg/mL of C1Mab-2, C1Mab-6, or an anti-IDH1 mAb (clone RcMab-1), followed by incubation with horseradish peroxidase-conjugated anti-rat IgG (1:10,000; Sigma-Aldrich Corp., St. Louis, MO). Finally, protein bands were detected with ImmunoStar LD (FUJIFILM Wako Pure Chemical Corporation) using a Sayaca-Imager (DRC Co. Ltd., Tokyo, Japan).
Results
Determination of the epitopes of anti-mCCR1 mAbs (C1Mab-2 and C1Mab-6) by flow cytometry using chimeric proteins
Anti-mCCR1 mAbs, clone C1Mab-2 (rat IgG2b, lambda) and C1Mab-6 (rat IgG2b, kappa) 18 were previously established and are applicable for flow cytometry and Western blotting (http://www.med-tohoku-antibody.com/topics/001_paper_antibody_PDIS.htm). To investigate the binding epitopes of C1Mab-2 and C1Mab-6, we focused on four extracellular regions of mCCR1: the N-terminal region [amino acid (aa) 1–34], ECL1 (aa 92–107), ECL2 (aa 172–197), and ECL3 (aa 265–281). These regions of mCCR1 were replaced with the corresponding regions of mCCR5, which is highly homologous to mCCR1. As shown in Figure 1, plasmids encoding mCCR5 with these specific mCCR1 segments—mCCR1p1–34, mCCR1p92–107, mCCR1p172–197, and mCCR1p265–281—were created. The chimeric proteins were transiently expressed in CHO-K1 cells, and their reactivities to C1Mab-2 and C1Mab-6 were analyzed via flow cytometry (Fig. 2A). Both C1Mab-2 and C1Mab-6 reacted with mCCR5 containing mCCR1p1–34 but not with those containing mCCR1p92–107, mCCR1p172–197, or mCCR1p265–281 (Fig. 2A and B). The cell-surface expression of each mutant was confirmed using anti-mCCR5 mAbs, C5Mab-2, and C5Mab-4, which recognize the mCCR5 ECL2 and N-terminal regions, respectively (Fig. 2C and D). These results suggest that C1Mab-2 and C1Mab-6 recognize the N-terminal region of mCCR1.
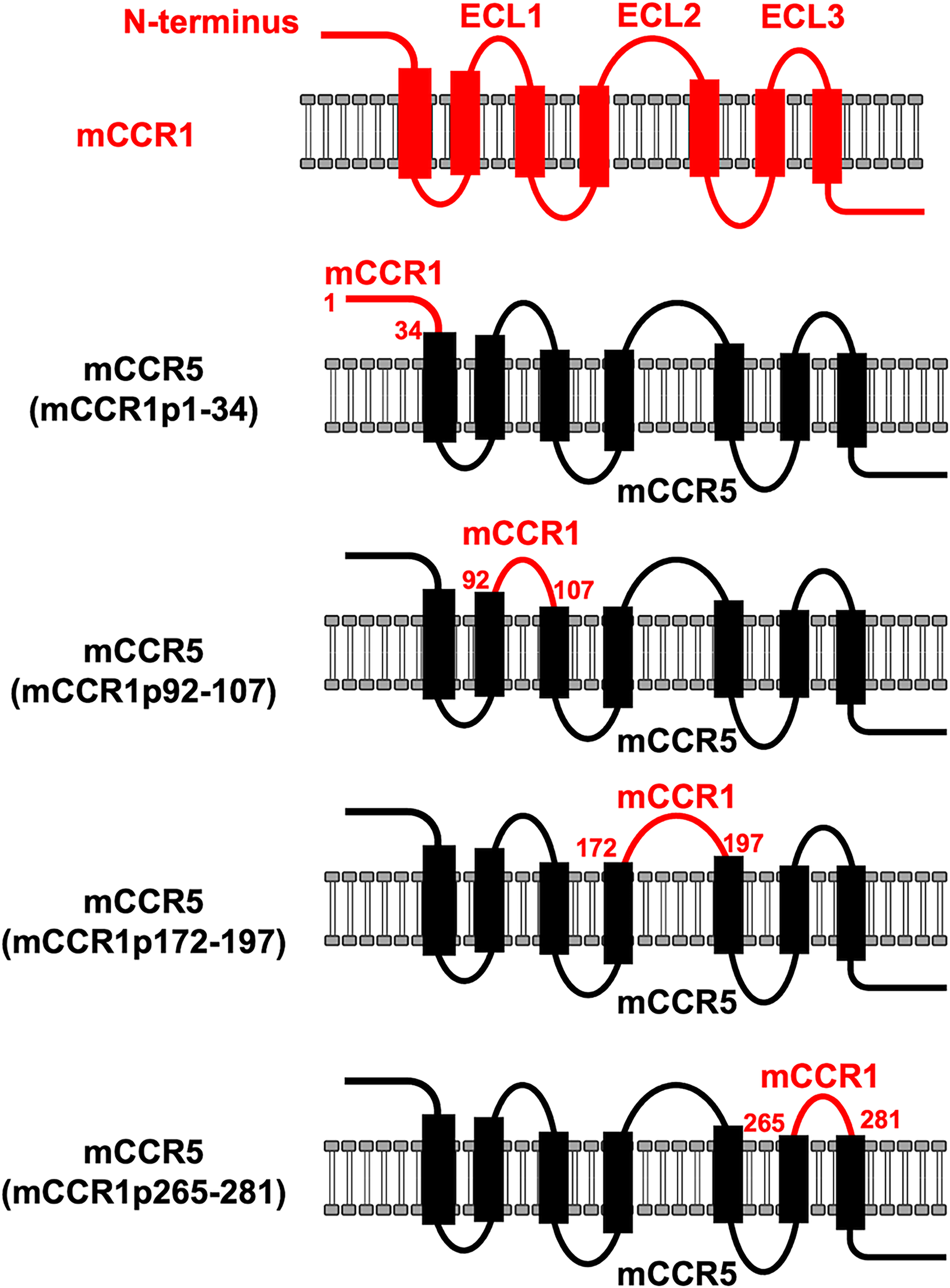
Schematic illustration of chimeric proteins. The four extracellular regions of mCCR1, including the N-terminal region (aa 1–34), ECL1 (aa 92–107), ECL2 (aa 172–197), and ECL3 (aa 265–281), were substituted into the corresponding regions of mCCR5. aa, amino acid; ECL, extracellular loop.
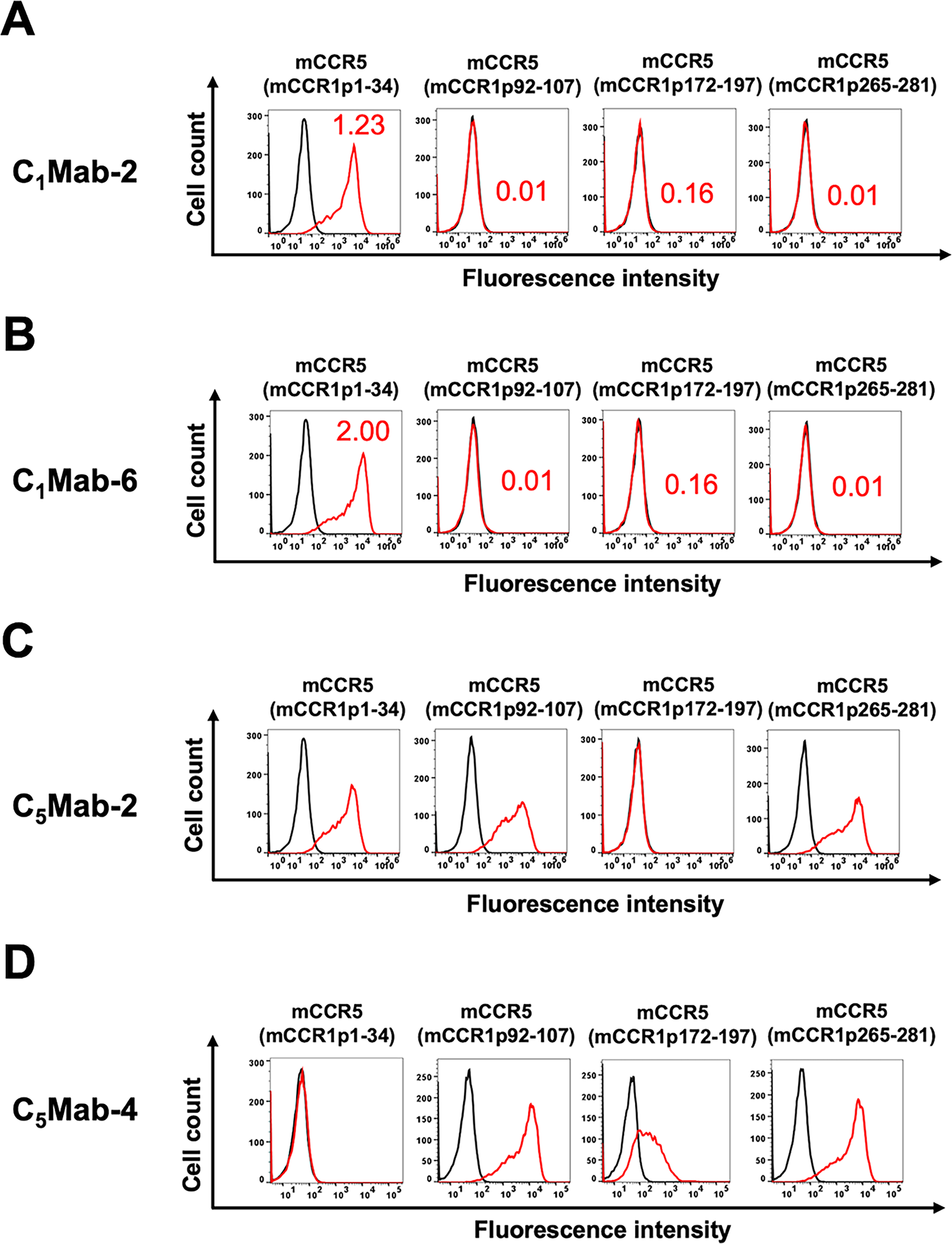
Determination of the epitope of anti-mCCR1 mAbs, C1Mab-2, and C1Mab-6 by flow cytometry using chimeric proteins. CHO-K1 cells transiently expressing the chimeric proteins were treated with C1Mab-2 (1 µg/mL,
Determination of the C1Mab-2 and C1Mab-6 epitopes by flow cytometry using PA-tag substitution
Next, we constructed PA-tag-substituted mCCR1 in the N-terminal region. As shown in Figure 3, PA-tag-substituted mCCR1 mutants on amino acids 2–13 (mCCR1-2PA13), 12–23 (mCCR1-12PA23), and 23–34 (mCCR1-23PA34) were created. Additionally, the 1st Met-substituted mCCR1 by PA-tag [PA-mCCR1 (2–355)] was produced. The PA-tag-substituted mCCR1 mutants were transiently expressed in CHO-K1 cells, and their reactivity to C1Mab-2 and C1Mab-6 was analyzed using flow cytometry. As shown in Figure 4, neither C1Mab-2 nor C1Mab-6 reacted with mCCR1-2PA13 nor PA-mCCR1 (2–355), but both reacted with mCCR1-12PA23 and mCCR1-23PA34. These results suggest that the epitopes of C1Mab-2 nor C1Mab-6 are located in the 1st Met and amino acids 2–13 of mCCR1.
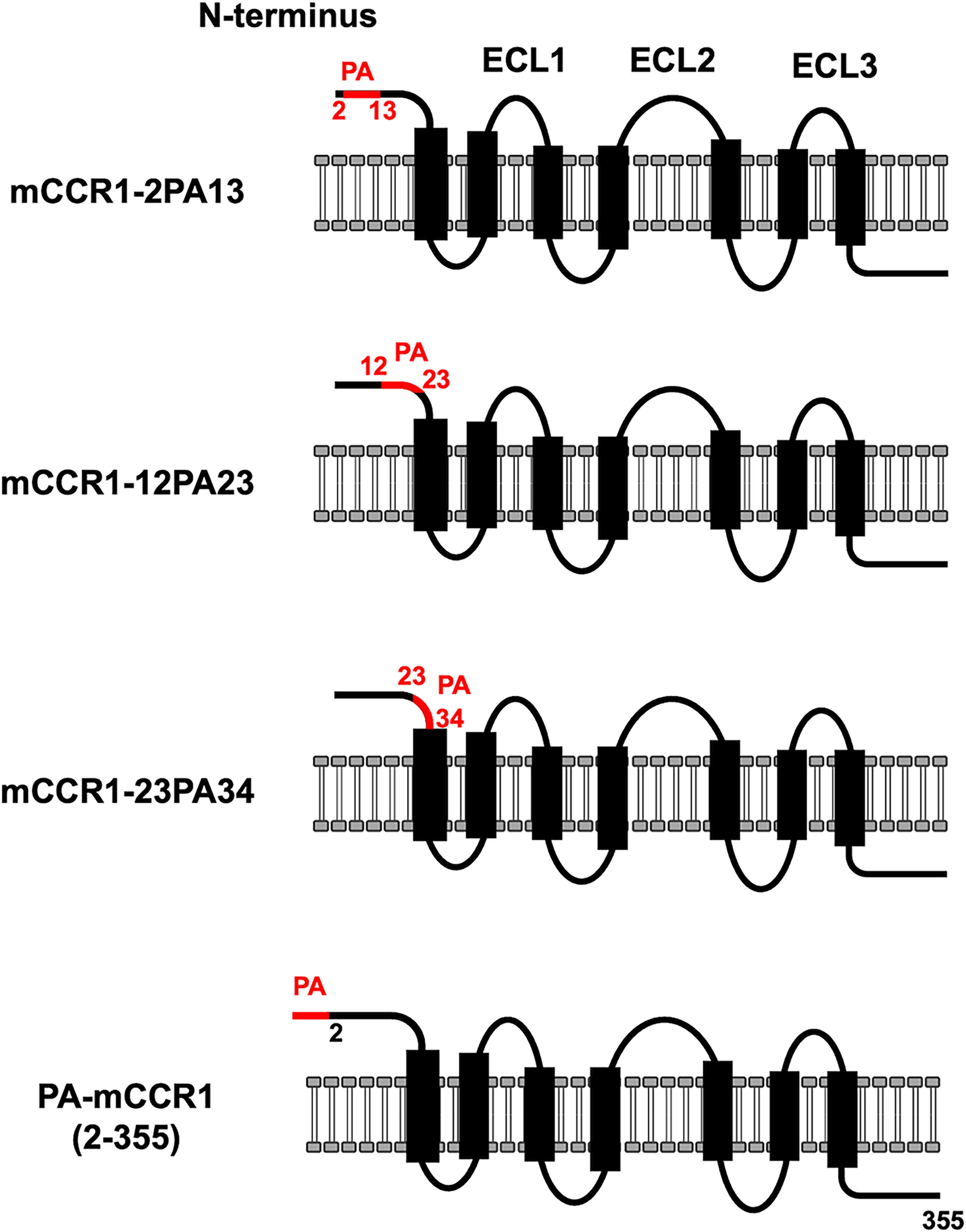
The illustration of PA-tag-substituted mutants of mCCR1.
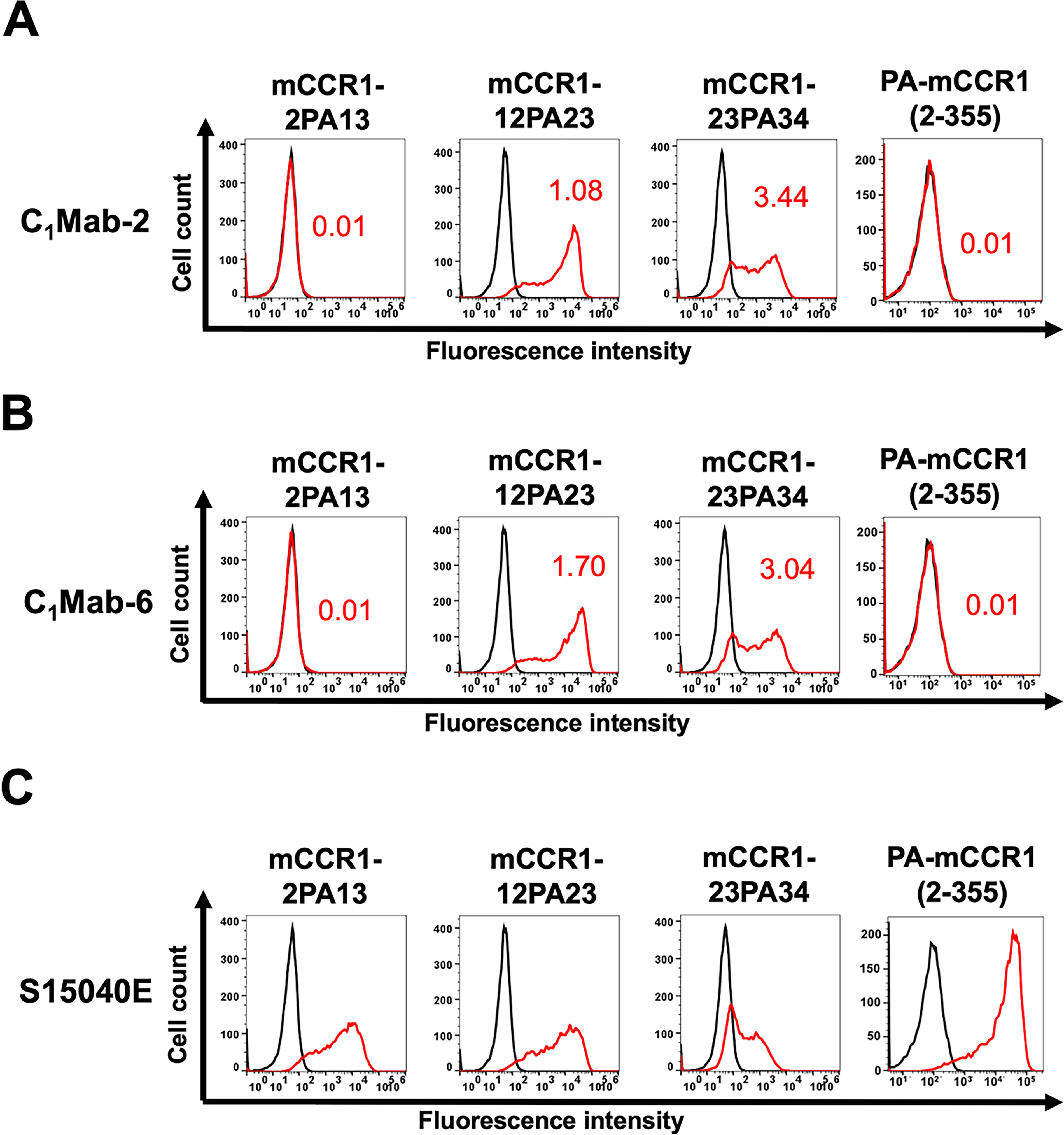
Determination of the C1Mab-2 and C1Mab-6 epitopes by flow cytometry using PA-tag-substituted mutants of mCCR1. CHO-K1 cells transiently expressing PA-tag-substituted mCCR1 mutants were treated with C1Mab-2 (1 µg/mL,
Determination of the C1Mab-2 and C1Mab-6 epitopes by flow cytometry using alanine scanning
Next, alanine scanning was performed on the N-terminal region (aa 2–13) of mCCR1. Twelve mutants with alanine (or glycine) substitutions in mCCR1 were created (Fig. 5), and these mutant proteins were transiently expressed in CHO-K1 cells. Reactivity with C1Mab-2 and C1Mab-6 was assessed by flow cytometry. As shown in Figure 6A, C1Mab-2 did not react with two mutants (E2A and F6A), and the reactivity of C1Mab-2 to the D5A mutant was reduced. Similarly, C1Mab-6 did not react with two mutants (E2A and D5A), and the reactivity of C1Mab-6 to the I3A mutant was reduced. (Fig. 6B). The cell surface expression of each mutant was confirmed with S15040E, which recognizes ECL2 (Fig. 6C). 19 The normalized reactivity of C1Mab-2 and C1Mab-6 (Fig. 6D) confirmed that Glu2, Asp5, and Phe6 are essential for recognition by C1Mab-2, and Glu2, Ile3, and Asp5 are crucial for recognition by C1Mab-6 in flow cytometry.
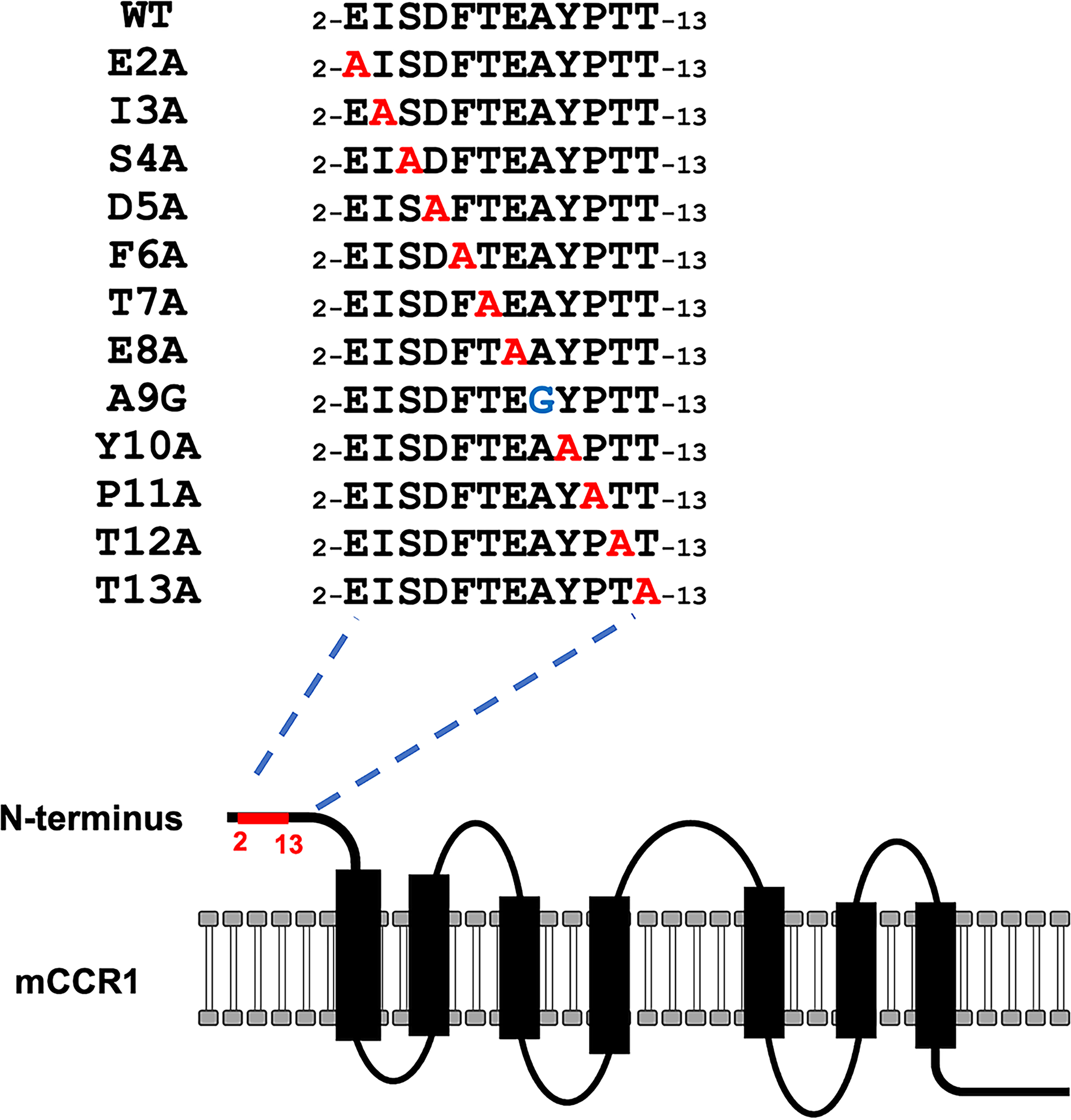
The illustration of alanine (or glycine)-substituted mutants of mCCR1.
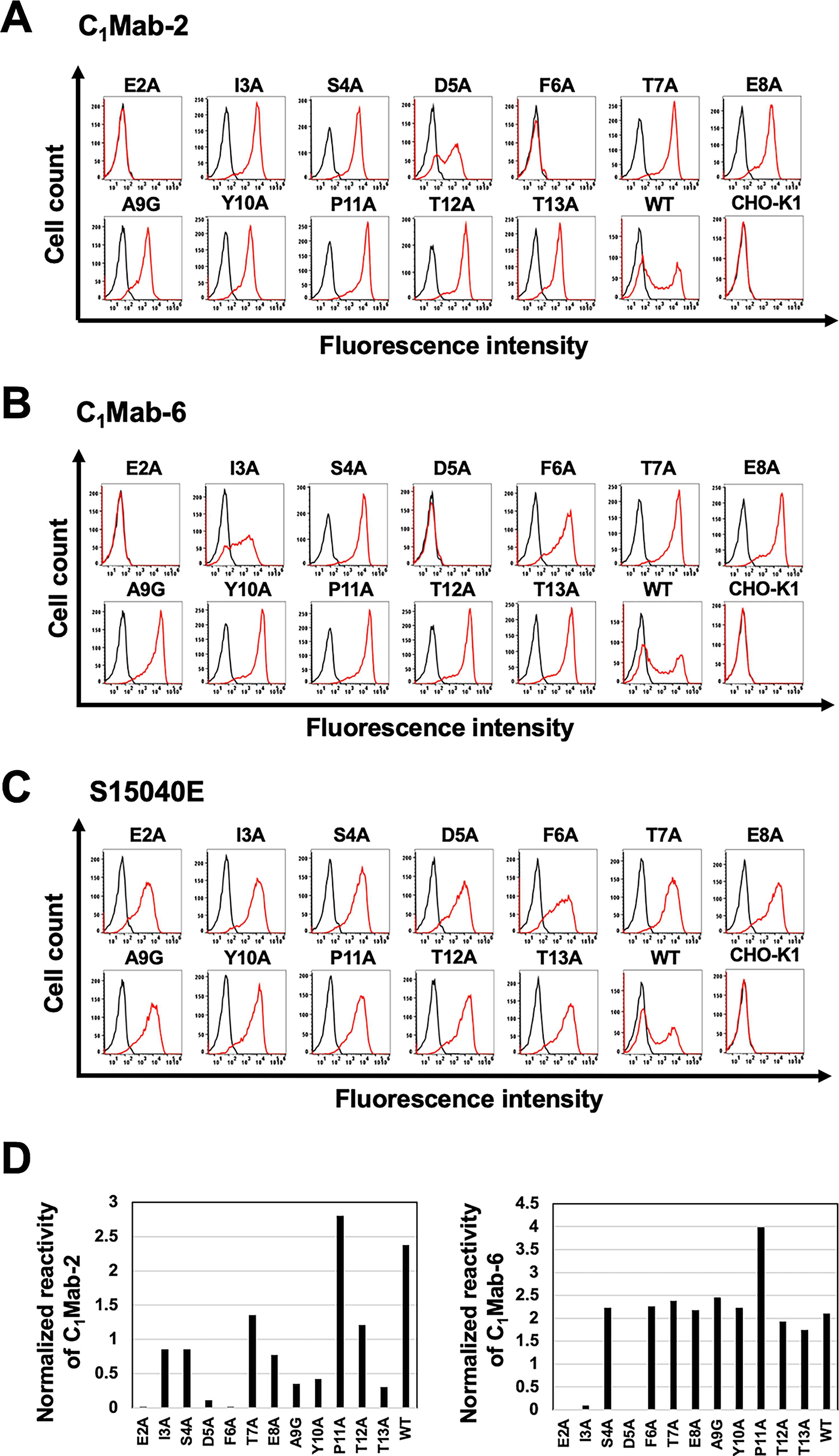
Determination of C1Mab-2 and C1Mab-6 epitopes by flow cytometry using alanine scanning. CHO-K1 transiently expressed mCCR1 mutants and wild-type (WT) were treated with C1Mab-2 (1 µg/mL,
Determination of the C1Mab-2 and C1Mab-6 epitopes by Western blotting using alanine scanning
Since C1Mab-2 and C1Mab-6 are suitable for Western blotting, the lysates of mutants were also examined using Western blotting. As shown in Figure 7A, C1Mab-2 detected 45–48 kDa and slower-migrating smear bands, which completely disappeared in the lysates of three mutants (E2A, D5A, and F6A). Furthermore, C1Mab-6 detected 45–48 kDa and slower migrating smear bands, which completely disappeared in the lysates of three mutants (E2A, I3A, and D5A; Fig. 7B). An anti-IDH1mAb, RcMab-1, was used as an internal control (Fig. 7C). These results were consistent with that of flow cytometry.
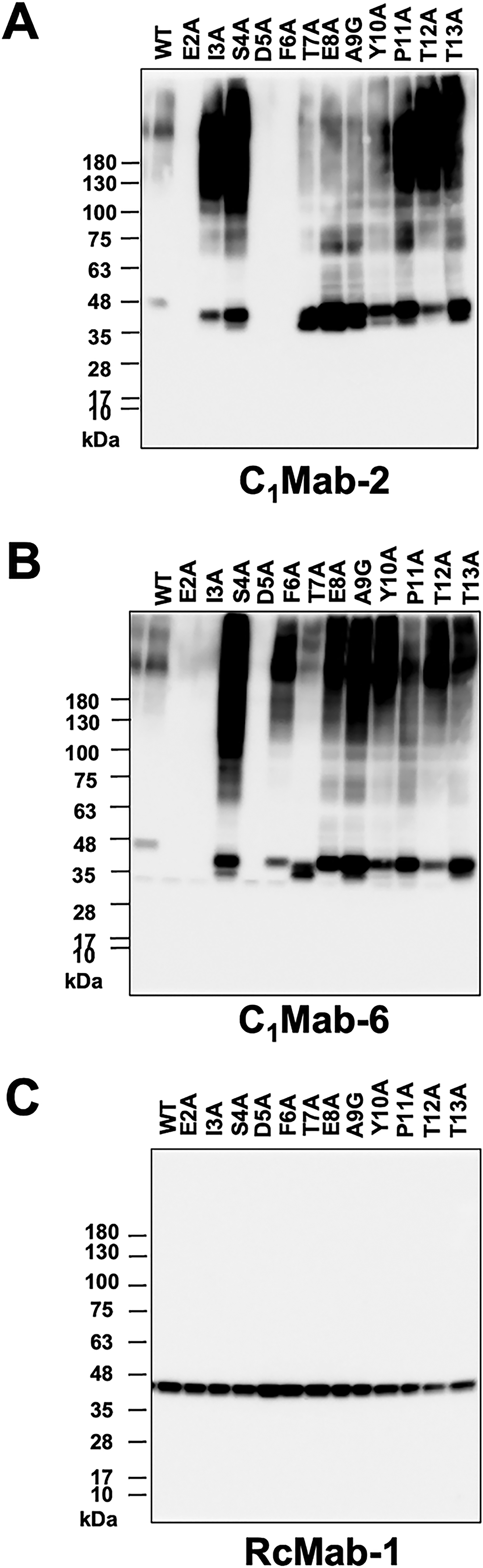
Determination of C1Mab-2 and C1Mab-6 epitopes by Western blotting. Cell lysates (10 μg/lane) from CHO-K1 cells transiently expressing mCCR1 mutants and wild-type (WT) were electrophoresed and transferred onto polyvinylidene difluoride membranes. The membranes were incubated with C1Mab-2 (1 μg/mL,
Discussion
This study conducted flow cytometry-based epitope mapping of anti-mCCR1 mAbs (C1Mab-2 and C1Mab-6) using chimeric proteins (Figs. 1 and 2) and PA-tag-substituted mutants (Figs. 3 and 4). Subsequently, alanine scanning revealed that Glu2, Asp5, and Phe6 are essential for recognition by C1Mab-2 in flow cytometry (Fig. 6A) and Western blotting (Fig. 7A). For C1Mab-6, Glu2, Ile3, and Asp5 are vital in flow cytometry (Fig. 6B) and Western blotting (Fig. 7B). Additionally, we found that C1Mab-2 and C1Mab-6 did not recognize the 1st Met-substituted mCCR1 [PA-mCCR1 (2–355)] in flow cytometry (Fig. 4), indicating that the 1st Met is also essential for the epitope. Figure 8 summarizes the results and illustrates the complementarity-determining regions (CDRs) of C1Mab-2 and C1Mab-6. Although C1Mab-2 and C1Mab-6 have different CDR sequences, they recognize similar epitopes in the mCCR1 N-terminal region.
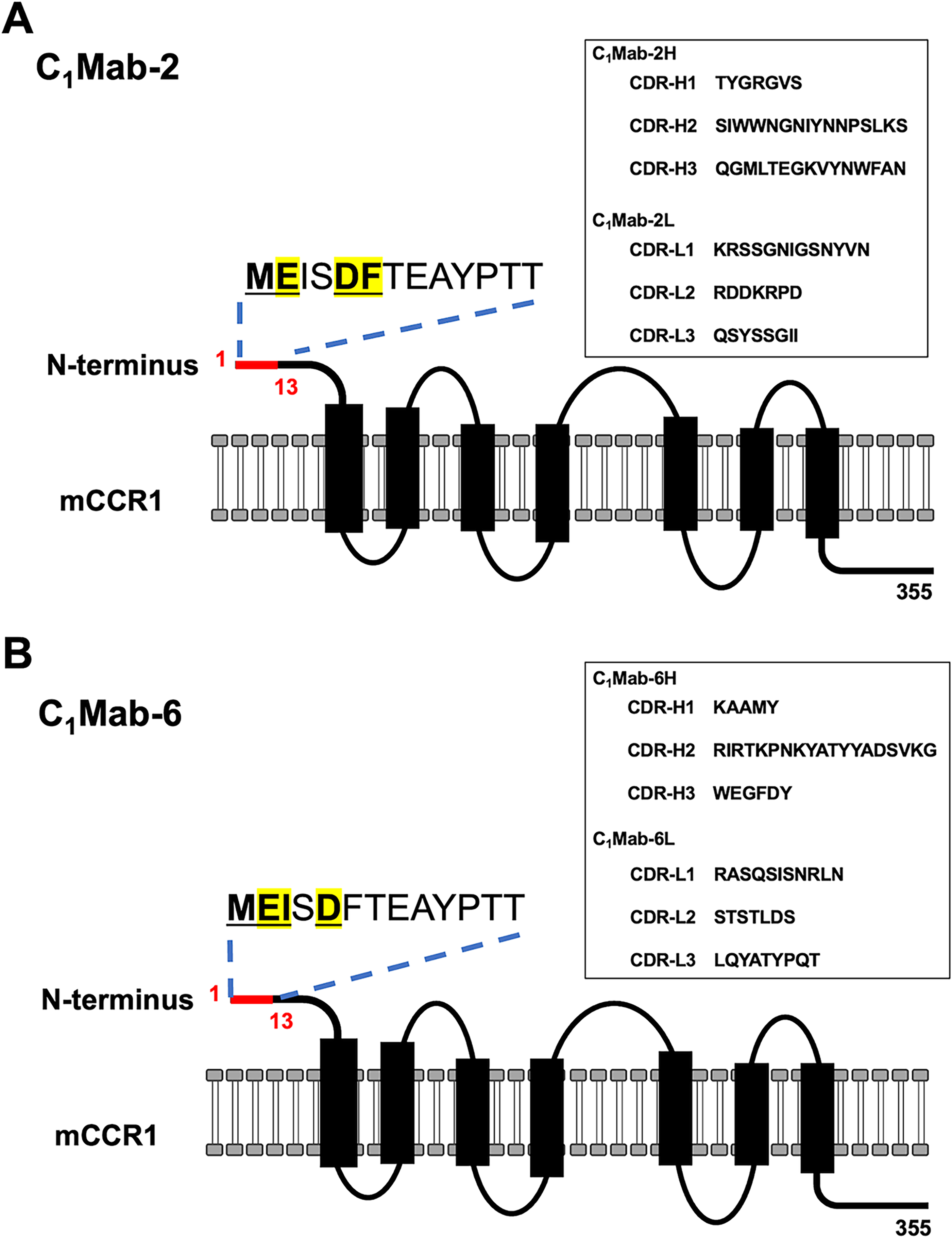
The schematic illustration of C1Mab-2 and C1Mab-6 epitopes.
The N-terminal region of the chemokine receptor is known as chemokine receptor site 1 (CRS1), which mainly mediates interactions with the chemokine N-loop/β3-strands and 40-second loop. These contacts are crucial for establishing the initial binding affinity and specificity between the chemokine and its cognate receptor. 22 CRS1 adopts a flexible and unstructured conformation when not in the presence of ligands.23–28 The identification of the epitope would contribute to the understanding of mAb-epitope interaction.
Chemokine–receptor interaction is a multistep process generally described by the two-site, two-step model within CRS1 and CRS2. In this model, chemokine–CRS1 interactions are initially established and facilitate ligand binding in the absence of receptor activation. After the initial docking at CRS1, a second interaction occurs at CRS2, where the chemokine N-terminus engages the deeper orthosteric binding pocket of the receptor. This subsequent interaction is essential for receptor activation, triggering the conformational changes needed for downstream intracellular processes signaling. 22 Since human CCR1 (hCCR1) N-terminal peptide (aa 1–29) serves as CRS1 to interact with the ligands, 29 corresponding mCCR1 N-terminus is thought to be involved in the ligand binding. Therefore, C1Mab-2 and C1Mab-6 are expected to neutralize the ligand binding. Further binding and functional studies are essential to prove the neutralizing effects.
CCR1 has been demonstrated as a drug target for the treatment of autoimmune diseases and tumors. 6 Recently, blocking mCCR1 with a neutralizing anti-CCR1 mAb (clone KM5908) in protumorigenic myeloid cells demonstrated therapeutic efficacy in a mouse syngeneic model of colorectal cancer. 30 KM5908 was established by immunizing Ccr1 knockout mice with cells overexpressed hCCR1 and recognized both hCCR1 and mCCR1. 31 Although KM5908 was selected based on its inhibitory activity on the chemotaxis of the THP-1 monocytic cell line toward CCL15 gradient, 31 the binding epitope has not been clarified. Our epitope mapping system will help identify the KM5908 epitope, which is important for comparing properties with C1Mab-2 and C1Mab-6.
Authors’ Contributions
A.O. performed the experiments. M.K.K. and Y.K. designed the experiments. H.S. and Y.K. wrote the article. All authors have read and agreed to the published version of the article.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest.
Funding Information
This research was supported in part by the Japan Agency for Medical Research and Development (AMED) under grant numbers: JP26am0521010 (to Y.K.), JP26ama121008 (to Y.K.), JP25ama221153 (to Y.K.), JP25ama221339 (to Y.K.), and JP25bm1123027 (to Y.K.) and by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) grant no. 25K10553 (to Y.K.).