
Abstract
Keywords
Introduction
Paclitaxel, a taxane chemotherapeutic agent that inhibits microtubule depolymerization, has been a therapeutic mainstay in the management of breast cancer, ovarian cancer, and other solid tumors. 1 Ocular adverse events occur in approximately 10% of treated patients and may range from mild symptoms such as dry eye, conjunctivitis, and epiphora to more severe complications such as optic neuropathy and cystoid macular edema (CME). 2 The incidence and underlying mechanism of macular edema in paclitaxel-associated maculopathy remain unclear. Proposed explanations include dysfunction of the retinal pigment epithelium (RPE) related to altered microtubule dynamics 3 and Müller cell toxicity,4,5 both of which are consistent with the characteristic absence of leakage on fluorescein angiography (FA).
CME is also a well-documented cause of vision loss in patients with inherited retinal diseases (IRDs), including retinitis pigmentosa, 6 X-linked retinoschisis, 7 gyrate atrophy, 8 NR2E3-related retinopathies, and choroideremia. 9 A large retinitis pigmentosa case series reported CME in approximately 25% of patients,10 –13 with angiographically silent CME accounting for one-third of such cases. 14 The proposed pathophysiology of CME in IRDs is multifactorial, with most hypotheses implicating vitreous traction, 15 defects in cell–cell adhesion, 16 and blood-retinal barrier impairment.17,18 Treatments for IRD-associated CME include topical and systemic carbonic anhydrase inhibitors, steroid and nonsteroidal anti-inflammatory drugs, and intravitreal antivascular endothelial growth factor (anti-VEGF) agents, with variable reported efficacy.19 –23
In this series, we report 3 cases of paclitaxel-associated CME with clinical features suggestive of an underlying IRD. In 1 patient, genetic testing confirmed a causative mutation consistent with enhanced S-cone syndrome. Another had several variants of uncertain significance affecting the LRP2 and RBP3 genes. The third patient was a carrier of a pathogenic mutation in PEX1, a gene associated with retinal degeneration in the setting of peroxisome biogenesis disorders.
Case Reports
Case 1
A 38-year-old woman with stage IIIC serous peritoneal carcinoma undergoing carboplatin and paclitaxel therapy for the preceding 2.5 months presented with blurry vision in both eyes. Her best-corrected visual acuity (BCVA) was 20/40 OD and 20/50 OS. Slitlamp examination revealed rare vitreous cells. Dilated fundus examination demonstrated CME and midperipheral hyper- and hypopigmentation in both eyes and an atrophic chorioretinal scar in the left eye (Figure 1). Optical coherence tomography (OCT) of the macula showed cystic intraretinal fluid bilaterally. Fundus autofluorescence revealed a bullseye pattern of perifoveal hyperautofluorescence in both eyes. FA exhibited peripheral nonperfusion and peripheral vascular leakage bilaterally, without leakage in the macula. An infectious and inflammatory workup, including syphilis screen, QuantiFERON-TB Gold testing, antineutrophil cytoplasmic antibody, and computed tomography of the chest, was unremarkable. Owing to the atypical pigmentary changes in both eyes, genetic testing for an underlying IRD was pursued, which identified a homozygous pathogenic NR2E3L mutation in the gene c.119-2A>C, suggestive of enhanced S-cone syndrome.
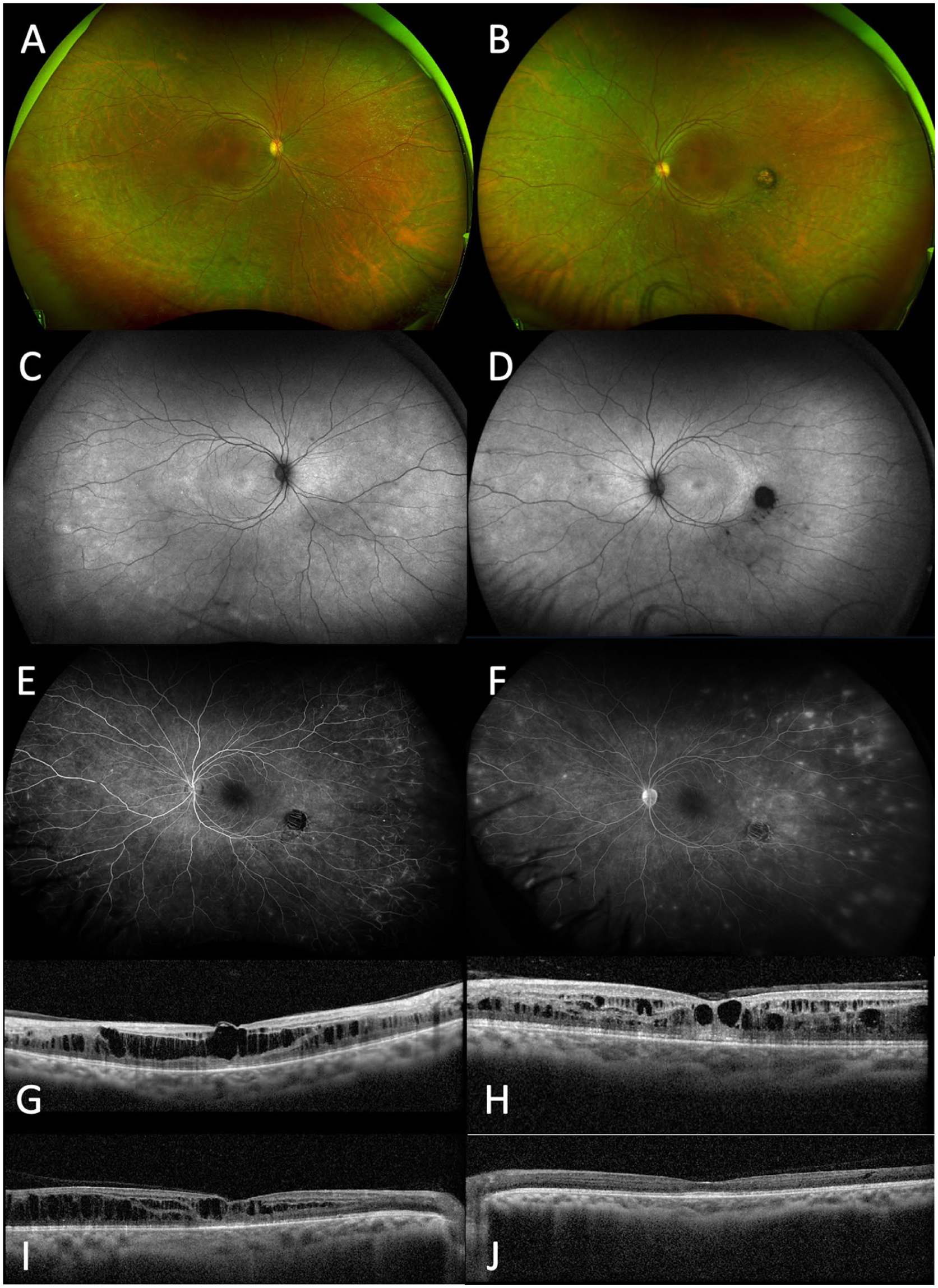
Case 1. Widefield color fundus photos of the (A) right and (B) left eyes show midperipheral hyper- and hypopigmentation bilaterally, with an atrophic chorioretinal scar in the left eye. Fundus autofluorescence of the (C) right and (D) left eye demonstrates a bullseye pattern of perifoveal hyperautofluorescence. Fluorescein angiography of the left eye reveals peripheral nonperfusion and multiple foci of peripheral vascular leakage in (E) early (0:54) and (F) late (9:30) frames. Optical coherence tomography (OCT) of the macula at initial presentation in the (G) right and (H) left eyes shows cystic intraretinal fluid, corresponding to visual acuities (VAs) of 20/40 and 20/50, respectively. OCT at the last follow-up of the (I) right and (J) left eyes demonstrates improvement of cystoid macular edema in the right eye and complete resolution in the left eye, with corresponding VAs of 20/40 and 20/30, respectively.
After discussion with the patient’s oncologist, paclitaxel, suspected to be the underlying cause of her CME, was discontinued. Topical corticosteroids, nonsteroidal anti-inflammatory drugs, and carbonic anhydrase inhibitors were trialed without clear benefit. Two months after discontinuation of paclitaxel, her BCVA improved to 20/40 OD and 20/30 OS, accompanied by a reduction in CME in the right eye and complete resolution in the left eye.
Case 2
A 51-year-old woman with metastatic breast cancer presented with blurry vision and occasional flashes in both eyes for 1.5 months. She had been previously treated with radiation and multiple chemotherapeutic agents, including paclitaxel, for 6 months before presentation. Paclitaxel had been discontinued by her oncologist 1 month after the development of blurry vision, before her referral to our clinic. On review of systems, she reported challenges with night vision since childhood, which had also affected her brother, mother, and aunt. Her BCVA was 20/50 OD and 20/70 OS. Slitlamp examination was unremarkable. Dilated fundus examination revealed CME, midperipheral RPE atrophy with pigment clumping, and peripheral vascular attenuation (Figure 2). Goldmann visual fields demonstrated moderate constriction of the I4e and I2e isopters in both eyes. OCT showed intraretinal cysts, and FA demonstrated no associated macular leakage. Although paclitaxel maculopathy was initially suspected, the peripheral chorioretinal atrophy and pigmentary changes were suggestive of an underlying IRD. The patient later denied difficulty with night vision or peripheral vision loss and any family history of ocular conditions. Genetic testing demonstrated 4 heterozygous mutations deemed to be variants of uncertain significance affecting the LRP2 and RBP3 genes.
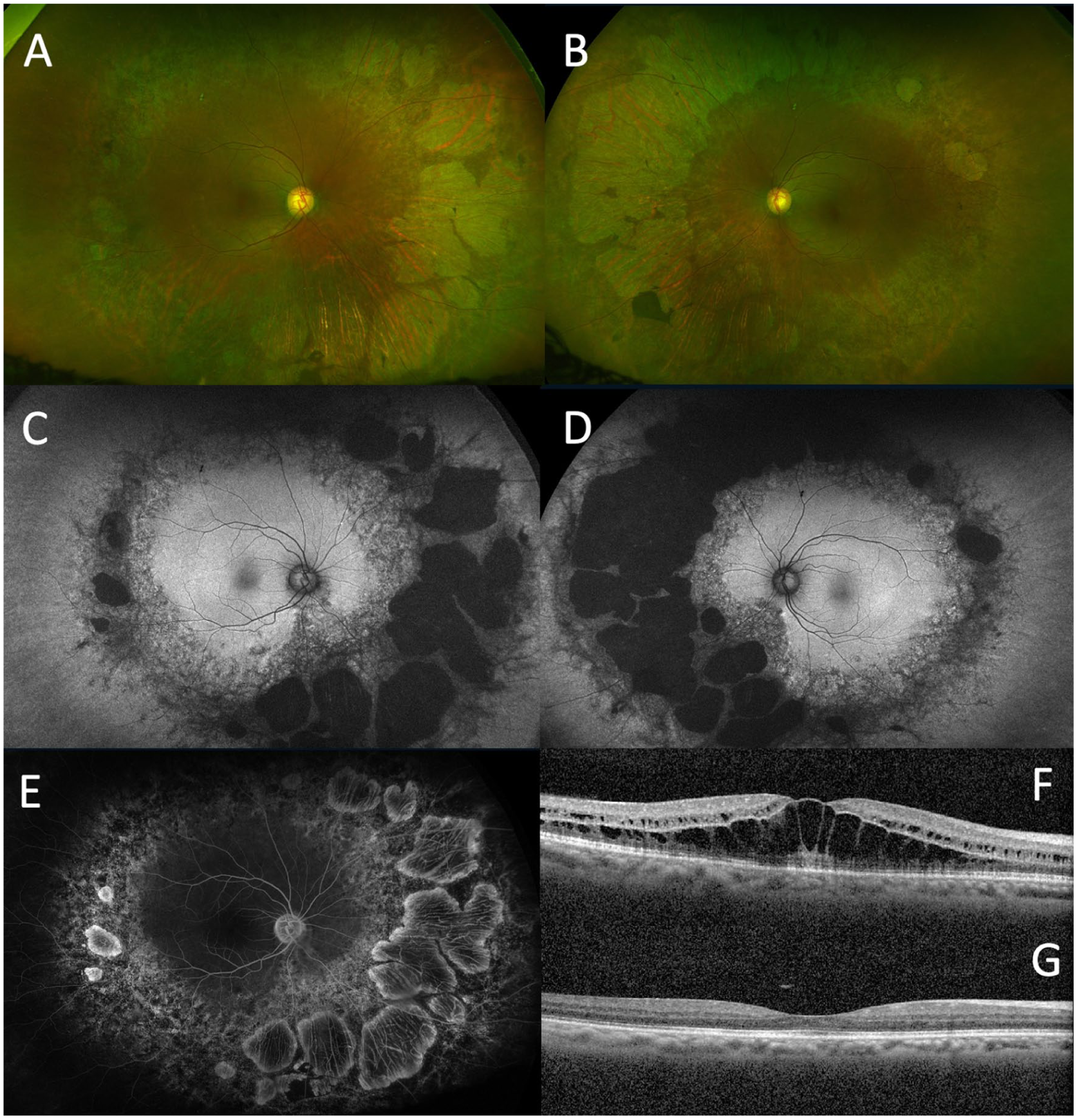
Case 2. Widefield color fundus photographs of the (A) right and (B) left eyes show midperipheral retinal pigment epithelial atrophy, pigment clumping, and vascular attenuation. Fundus autofluorescence of the (C) right and (D) left eye demonstrates midperipheral hypoautofluorescence and adjacent speckled hyperautofluorescence in the near periphery. (E) Fluorescein angiography of the right eye shows no late macular leakage. Optical coherence tomography macula of the right eye at (F) initial presentation and (G) final follow-up shows resolution of cystoid macular edema and improvement in visual acuity from 20/50 to 20/30. Similar findings were noted in the left eye.
The patient was started on topical steroids in both eyes. About 1 month after initiation of treatment, and 3 months after discontinuation of paclitaxel, the CME had resolved, and her vision subjectively returned to baseline, with BCVA improving to 20/30 OD and 20/40 OS.
Case 3
A 68-year-old man with a history of metastatic oral squamous cell carcinoma presented with progressively worsening bilateral vision 7 weeks after initiation of a combination therapy with paclitaxel, carboplatin, and pembrolizumab. His medical history was otherwise unremarkable, and his ocular history was notable for a childhood clinical diagnosis of retinitis pigmentosa, although he denied having undergone an ophthalmic examination for several decades or having any prior functional limitations related to his vision. There was no known family history of IRD. His BCVA was 20/80 OD and 20/50 OS. Dilated fundus examination revealed bilateral CME, which was more pronounced in the right eye, along with symmetric confluent nummular areas of RPE atrophy and pigment clumping in the midperiphery of both eyes (Figure 3). FA demonstrated no macular leakage. There was no evidence of intraocular inflammation to suggest immune checkpoint inhibitor-associated uveitis. Genetic testing revealed a heterozygous pathogenic PEX1 c.2097dup variant, previously associated with autosomal recessive peroxisome biogenesis disorders associated with retinal degeneration.
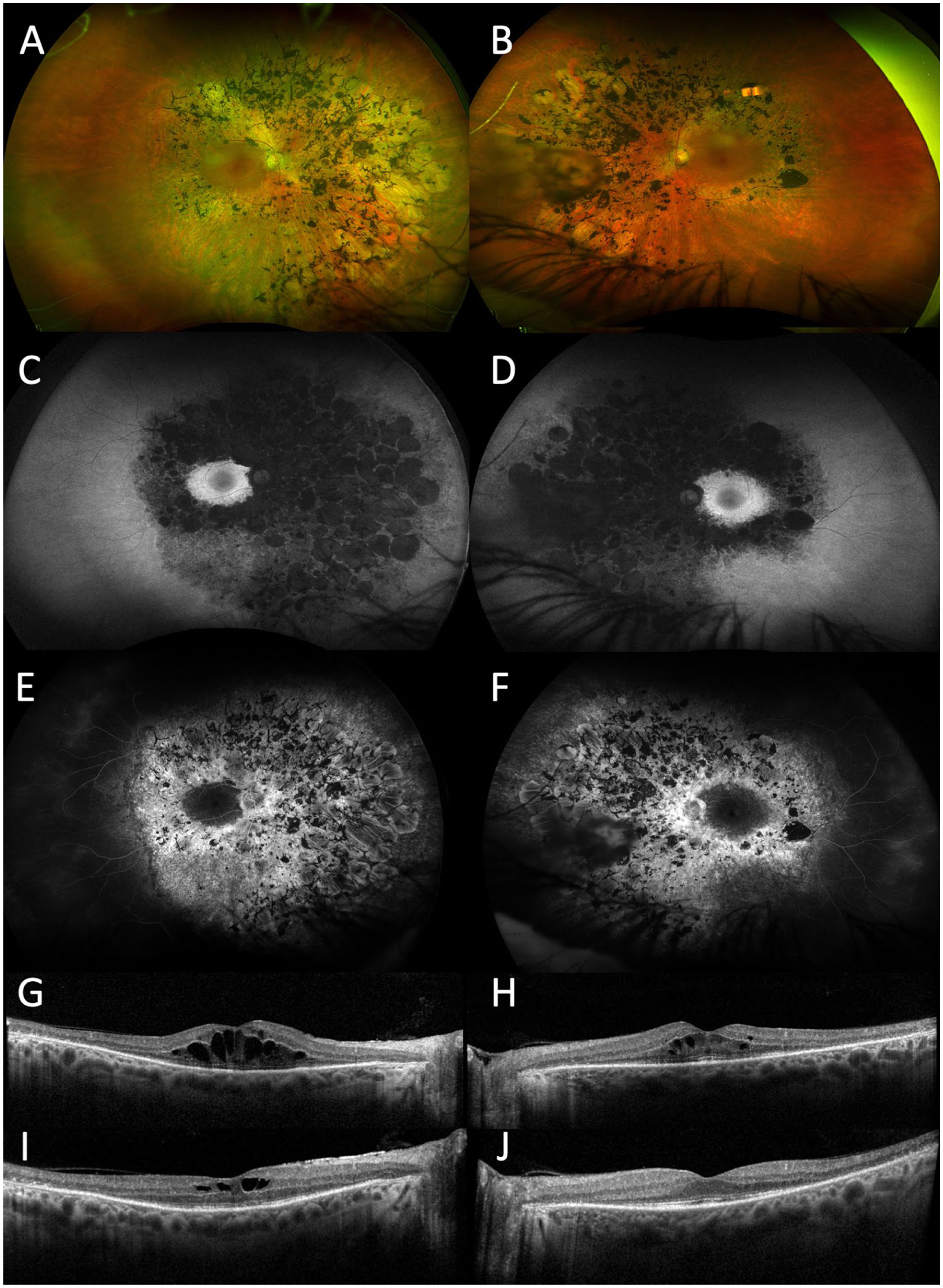
Case 3. Widefield color fundus photographs of the (A) right and (B) left eyes show symmetric, confluent nummular areas of retinal pigment epithelial atrophy and pigment clumping in the midperipheral retina bilaterally. Fundus autofluorescence of the (C) right and (D) left eyes demonstrates corresponding midperipheral hypoautofluorescence sparing the macula. Fluorescein angiography of the (E) right and (F) left eyes shows no late macular leakage. Optical coherence tomography (OCT) of the macula at initial presentation in the (G) right and (H) left eyes reveals cystoid macular edema (CME), corresponding to visual acuities of 20/80 and 20/50, respectively. OCT at the last follow-up of the (I) right and (J) left eyes shows improvement of CME in the right eye and complete resolution in the left eye, with visual acuity improving to 20/50 and 20/30, respectively.
After discussion with his oncology team, both carboplatin and paclitaxel were discontinued. The patient was started on topical prednisolone, dorzolamide, and ketorolac, which were tapered gradually. Six months after initial presentation, his BCVA had improved to 20/50 OD with a significant reduction in macular edema and to 20/30 OS with complete resolution of macular edema.
Conclusions
In this series, we describe 3 patients with clinical features of IRD who developed CME after exposure to paclitaxel. In all cases, visual acuity and macular edema improved over several months after discontinuing paclitaxel, in conjunction with topical and systemic treatment. These observations suggest that IRD may be a risk factor for the development of paclitaxel-associated CME.
Paclitaxel-associated CME is typically bilateral, angiographically silent, and usually demonstrates spontaneous resolution after cessation of the medication.3,24 In a study of 1533 patients receiving paclitaxel chemotherapy, angiographically silent CME was found in 0.3%. 25 A similar angiographically silent CME has been observed in numerous IRDs, a disease feature increasingly recognized with the advent of OCT imaging. 12 While the exact cause of this angiographically silent CME is uncertain, proposed mechanisms in both IRD-associated CME as well as paclitaxel maculopathy involve Müller cell dysfunction.26 –28 Paclitaxel is suspected to disrupt Müller cell function, as demonstrated by reduced b-wave amplitude in electroretinography studies. 29 We therefore hypothesize that patients with IRDs may have an increased susceptibility to paclitaxel-associated CME owing to underlying structural and functional retinal vulnerability. Many IRDs are associated with chronic Müller cell stress; breakdown of the external limiting membrane; and disrupted interactions between photoreceptors, Müller cells, and the RPE.26 –28 Such structural abnormalities impair fluid regulation and homeostasis, which may predispose the retinal tissue to intraretinal fluid accumulation.30 –32 Paclitaxel, through its stabilizing effect on microtubules, interferes with intracellular trafficking in retinal glia and the RPE, further limiting the clearance of excess fluid and cellular maintenance.3,29 Superimposing this drug effect on an already compromised retinal environment may lower the threshold for the development of CME.
The 3 patients in this series developed visual symptoms after receiving paclitaxel for 2.5 months, 6 months, and 7 weeks, respectively, prompting presentation to an ophthalmologist. In a previously reported series of 14 patients with breast cancer diagnosed with paclitaxel maculopathy, the time to onset of bilateral CME ranged from 1.5 to 27 months, with a mean of approximately 9 months. 33 The onset of symptoms after paclitaxel initiation in our patients occurred earlier than the reported mean of 9 months, yet remained within the reported range. Whether this relatively shorter time to onset of CME reflects increased susceptibility related to underlying IRD remains unclear and cannot be meaningfully assessed owing to the limited sample size.
The patient in Case 1 in our series had a pathogenic homozygous mutation in the NR2E3L gene, c.119-2A>C, suggestive of enhanced S-cone syndrome. 34 Enhanced S-cone syndrome is an autosomal recessive retinopathy first clinically described in 1990. 34 Pathogenic variants in NR2E3, a photoreceptor-specific nuclear receptor,35,36 cause aberrant cellular differentiation and an overrepresentation of S-cones relative to other cone subtypes.37,38 The disease severity and rate of progression vary widely among patients. Common clinical features include intraretinal yellow-white lesions, nummular pigmentary changes, foveomacular schisis, and CME.34,39 In a large cohort of enhanced S-cone syndrome, foveomacular schisis was present in 41% of patients 40 and represented a significant source of visual decline irrespective of photoreceptor loss.40 –42 The associated macular edema is typically angiographically silent, 39 as observed in our patient, and treatment with topical and/or systemic carbonic-anhydrase inhibitors has shown mixed results, with therapeutic benefit presumed to arise from enhanced RPE pump function.43,44 The underlying mechanism of retinoschisis in enhanced S-cone syndrome remains unclear, though various theories implicate poor structural integrity due to insufficient tight junctions between aberrant photoreceptor inner segments and Müller cells. 45
In the second and third cases, there was no definitive genetic cause identified for IRD, but both demonstrated clinical features strongly suggestive of an underlying dystrophy, most notably pigmentary retinopathy. Although their genetic testing revealed either heterozygous pathogenic variants or variants of uncertain significance in IRD-associated genes, it is well recognized that current IRD gene panels may not identify causative mutations in up to 40% of clinically diagnosed cases.46,47 The second case was heterozygous for 4 genetic variants of uncertain significance: 3 in the LRP2 gene and 1 in the RBP3 gene. Both genes have been implicated in IRD. RBP3 encodes a large secreted protein that transports retinol between photoreceptors and the RPE. 48 Patients in the largest published series of RBP3-associated retinopathy also presented with CME and varying degrees of midperipheral pigmentation, although angiographic findings were not reported. 49
LRP2 variants are associated with Donnai–Barrow syndrome, which is classically characterized by craniofacial anomalies, underdeveloped corpus callosum, hereditary deafness, high myopia, and retinal dystrophy. 50 Our patient denied any hearing changes and did not exhibit high myopia or other systemic abnormalities consistent with this syndrome. In Case 3, genetic testing revealed a heterozygous pathogenic PEX1 variant, a gene known to cause rare autosomal recessive peroxisome biogenesis disorders such as Zellweger spectrum disorder and Refsum disease, both of which can feature retinal pigmentary dystrophy. To our knowledge, this is the first case of a heterozygous PEX1 loss-of-function mutation associated with ophthalmic findings; all previous reports of pigmentary retinopathy in PEX1-related disease have involved homozygous or compound heterozygous mutations. 51
These findings highlight the diagnostic challenges associated with variants of uncertain significance and heterozygous pathogenic mutations in genes associated with recessive IRDs. Although such findings do not meet the criteria for molecular confirmation of disease, they may still carry biological relevance, especially in the presence of suggestive clinical features. Variants of uncertain significance may represent variants with incomplete penetrance, undetected pathogenic variants on the second allele, or genetic modifiers that increase retinal vulnerability. Likewise, heterozygous carriers of recessive IRD genes, although usually asymptomatic, may demonstrate subtle retinal structural or functional abnormalities that lower the threshold for adverse responses to retinal stressors such as paclitaxel.
The limitations of current genetic testing underscore the importance of integrating molecular data with clinical findings. In the context of this report, these nuances support a cautious interpretation of the strength of association between IRD and paclitaxel maculopathy, while acknowledging the possibility of increased susceptibility in genetically predisposed individuals.
A review of each patient’s medications and medical history demonstrated no other associations or causes of pigmentary retinopathy. The patient in Case 2 had also been treated with atezolizumab and radiation therapy, neither of which has been reported to cause pigmentary retinopathy. In the third case, there was no significant medical history, and the patient was treated with carboplatin and pembrolizumab, neither of which has been associated with pigmentary retinopathy. 52 Pembrolizumab, an immune checkpoint inhibitor, has been associated with immune-related ocular adverse events, including uveitis, serous retinal detachment, and neuroretinopathy; however, there have been no reports to date linking immune checkpoint inhibitors to angiographically silent CME or pigmentary retinopathy. 53 Furthermore, this patient was diagnosed clinically with retinitis pigmentosa in childhood, suggesting that his pigmentary retinal changes were long-standing and not treatment-related.
Given the low incidence of paclitaxel maculopathy (~0.3%) and the rarity of IRDs, the observed overlap in these patients may be coincidental. However, the consistent clinical course across all 3 patients, with all demonstrating angiographically silent CME shortly after paclitaxel exposure and improvement following cessation, raises the possibility of an underlying susceptibility, warranting further investigation.
All 3 patients experienced improvement or resolution of macular edema within weeks to months after paclitaxel discontinuation. In a series of 14 patients with paclitaxel maculopathy, discontinuation of the drug in 12 cases led to visual improvement after an average of 2.7 months. 33 In that series, patients were treated with a variety of agents, including corticosteroids, nonsteroidal anti-inflammatory drugs, local and systemic carbonic anhydrase inhibitors, anti-VEGF injections, or simple observation, thereby limiting conclusions regarding the relative effectiveness of any specific treatment modality. Similarly, in our cohort, it is impossible to determine whether the improvement in our patients was related to medical intervention or was simply the result of paclitaxel discontinuation. These 3 patients were identified over a period of several years at 2 separate academic institutions, highlighting the rarity of this potential association and emphasizing the need for future case collection and validation.
In this series, we present 3 patients with paclitaxel maculopathy possibly related to underlying IRD and increased susceptibility to drug toxicity. Although larger studies are needed to validate this association, these findings suggest that patients initiating paclitaxel, particularly those with symptoms or a family history of IRD, may benefit from a baseline ophthalmic examination, including fundus photography and OCT imaging.
Footnotes
Ethical Approval
The authors’ institution does not require formal ethical approval for reporting individual patient cases.
Statement of Informed Consent
Verbal informed consent was obtained from all patients for inclusion in this case report.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Eliot is a consultant for Alcon, Aldeyra Therapeutics, Apellis, ExOcular, GelMedix, Genentech, Neurotech, Pykus Therapeutics, and RetMap; DSMB member for Asclepix, Aviceda, Clearside Biomedical, EyeBio, and Ocular Therapeutix; stockholder of Aldeyra Therapeutics, Cellio, ExOcular, InGel, Ingenia Therapeutics, Pykus Therapeutics, and RetMap; and received research grants from Neurotech and patent royalties from Aldeyra Therapeutics.
Dr. Yuan is a consultant for Ocular Therapeutix. The funding organizations had no role in the design or conduct of this research. None of the other authors declared potential conflicts of interest with respect to the research, authorship, and/or publication of the article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.