
Abstract
Introduction
A healthy human retina consists of approximately 5% cone photoreceptors, which are responsible for nearly all visual functions in daily life, including color vision. In contrast, rod photoreceptors, which account for about 95% of the retina, mediate vision under low-light conditions. 1 Embryologically, cone and rod cells differentiate from a common progenitor cell population. This differentiation is regulated by the coordinated action of multiple genes, among which NR2E3 (Nuclear Receptor Subfamily 2 Group E Member 3) plays a significant role. In addition, studies indicate that NR2E3 plays a critical role in retinal stability during adulthood. 2 Over 80 variants of NR2E3 associated with retinal diseases have been identified. These variants predominantly exhibit an autosomal recessive inheritance pattern and are associated with conditions such as enhanced S-cone syndrome, Goldmann–Favre syndrome, retinitis pigmentosa, and clumped pigmentary retinal degeneration. 2
Enhanced S-cone syndrome is a rare inherited retinal dystrophy thought to be caused by a mutation in the NR2E3 gene. These mutations result in a rod photoreceptor dysfunction due to a reduction in the number of rod cells or structural abnormalities, along with a reduction in M-cone and L-cone cells and an increase in S-cone cells. Enhanced S-cone syndrome typically presents with nyctalopia. The clinical presentation can vary widely; children may exhibit a normal fundus appearance, whereas adults more commonly demonstrate characteristic nummular pigmentary changes around the vascular arcades. Foveoschisis, also known as nonvasogenic cystoid maculopathy, can lead to decreased visual acuity and may accompany enhanced S-cone syndrome.3,4
In this report, we present the case of a 45-year-old man who presented with visual impairment and was found to have bilateral foveoschisis on multimodal imaging, with pigmentary changes limited to the peripheral retina. Genetic analysis revealed a homozygous mutation in the NR2E3 gene. These genetic findings and electrophysiologic test results led to a diagnosis of an atypical form of enhanced S-cone syndrome associated with foveoschisis.
Case Report
A 45-year-old man presented with a long-standing history of decreased vision in his left eye. The patient’s medical history revealed that the visual symptoms were not present during childhood or early adulthood but developed later in life. He denied nyctalopia, and there was no family history of similar ocular diseases. On examination, best-corrected visual acuity (BCVA) was 20/32 OD and 20/100 OS. Bilateral intraocular pressures and anterior segment examinations were within normal limits. Fundus examination revealed cystoid alterations in the fovea (Figure 1A). Additionally, yellow-white deposits accompanied by pigmentary changes were observed bilaterally in the peripheral retina (Figure 1B). Macular optical coherence tomography demonstrated bilateral foveoschisis (Figure 2). Fundus autofluorescence imaging demonstrated mild bilateral foveal abnormalities due to the cystoid macular changes, with an additional hypoautofluorescent area in the right fovea due to a pre-existing retinal pigment epithelium defect. Subsequent fundus fluorescein angiography demonstrated no leakage from the cystic spaces (Figure 3, A and B).
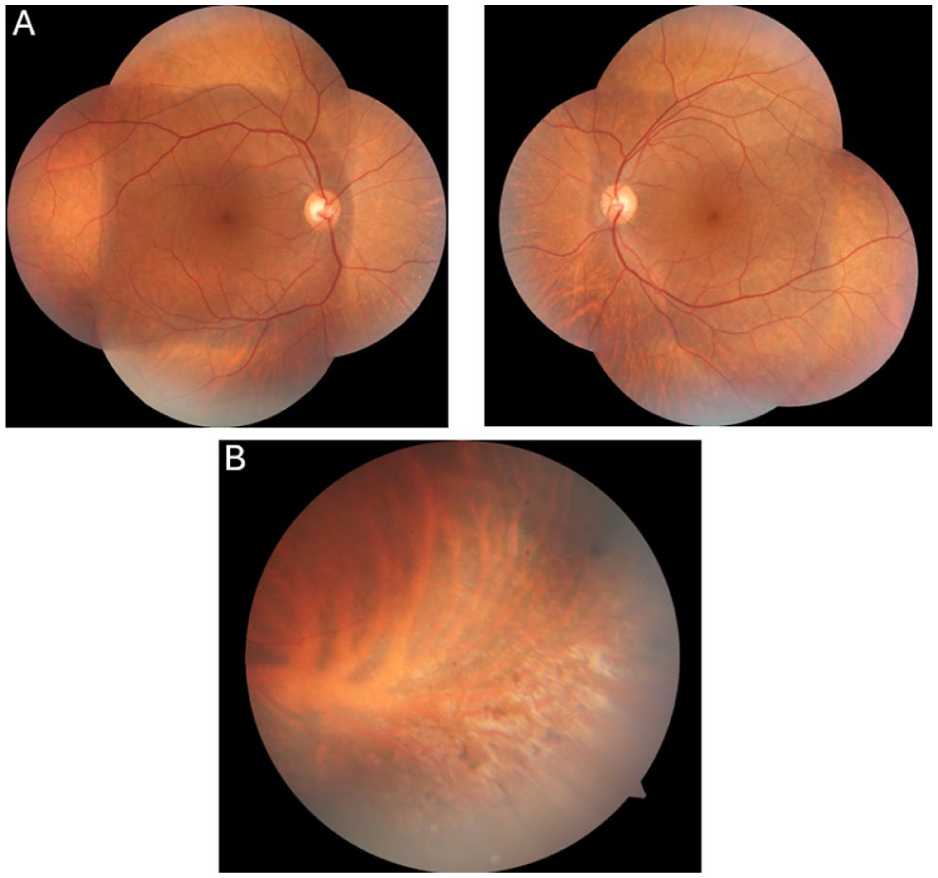
Color fundus images obtained at presentation showing (A) mild cystoid alterations in the fovea. (B) Peripheral retinal imaging of the left eye shows yellow-white dots with pigmentary hyperplastic changes.
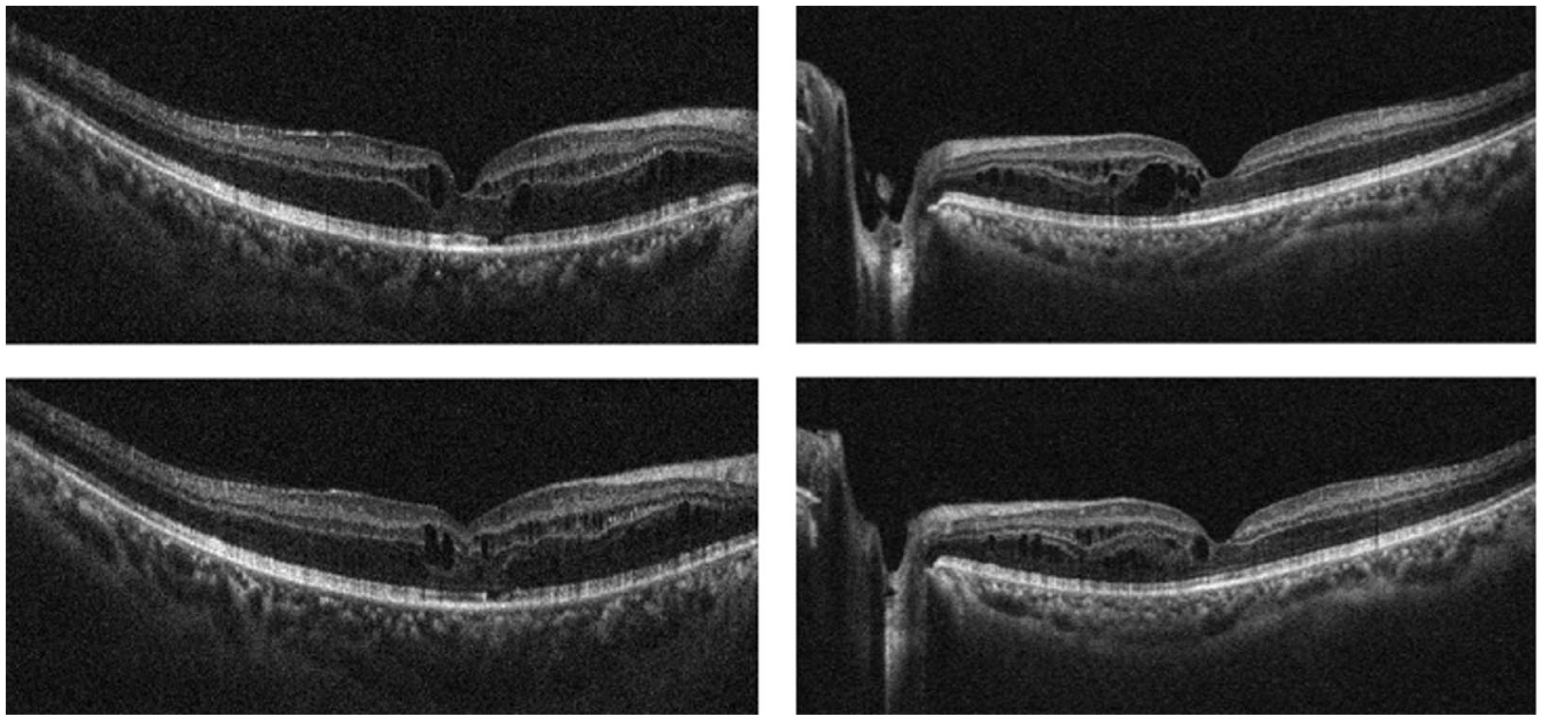
Optical coherence tomography images centered on the fovea at presentation demonstrating bilateral foveoschisis with intraretinal cystoid spaces in both eyes.
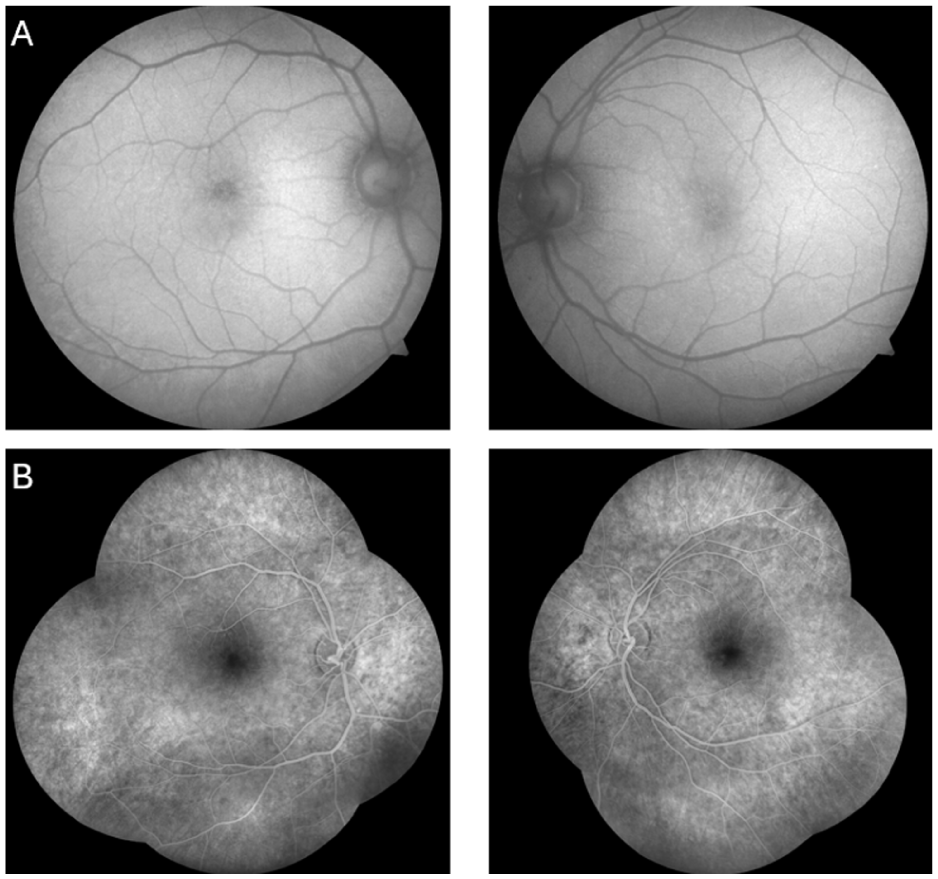
Fundus autofluorescence images obtained at presentation reveal bilateral foveal autofluorescence abnormalities, with a (A) hypoautoflourescent area corresponding to a retinal pigment epithelium defect in the right eye. (B) Fundus fluorescein angiography performed at the same visit demonstrates no leakage from the cystoid spaces. A window defect pattern is also observed superior to the fovea of the right eye.
The patient underwent blue-flash and full-field electroretinography (ERG) according to International Society for Clinical Electrophysiology of Vision protocols. In the bilateral dark-adapted 0.01 scotopic test, neither a-waves nor b-waves were detected. Significant reductions in the amplitudes of both a-waves and b-waves were observed in the dark-adapted 3.0 and dark-adapted 10.0 tests. Bilateral oscillatory responses were also decreased. In addition, the bilateral light-adapted 3.0 flicker responses demonstrated reduced amplitudes and delayed implicit times compared with healthy retinal responses (Figure 4). Blue-flash responses recorded on an orange background showed significantly increased amplitudes compared with those of healthy retinal responses (Figure 5).
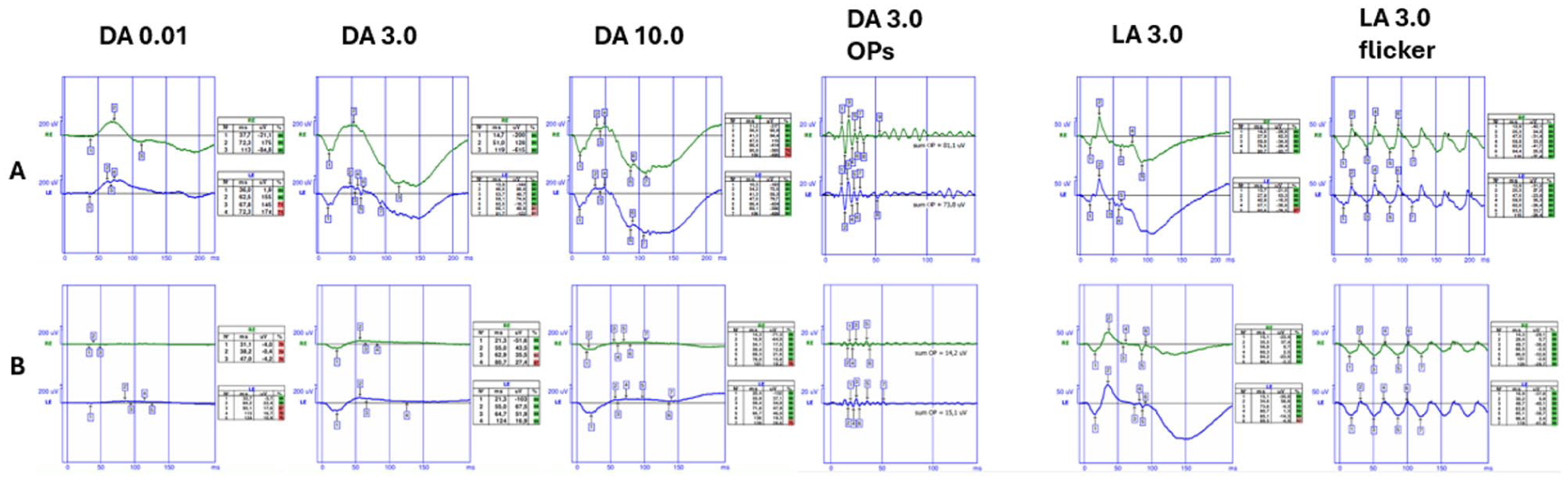
Full-field electroretinography findings of a healthy individual (row A) and the patient (row B). The patient demonstrates no a-wave or b-wave responses on dark-adapted 0.01 testing, low-voltage a and b waves on dark-adapted 3.0 and dark-adapted 10.0 testing, reduced amplitudes on dark-adapted 3.0 oscillatory potentials and light-adapted 3.0 responses, and delayed and low amplitudes on light-adapted 3.0 flicker responses.
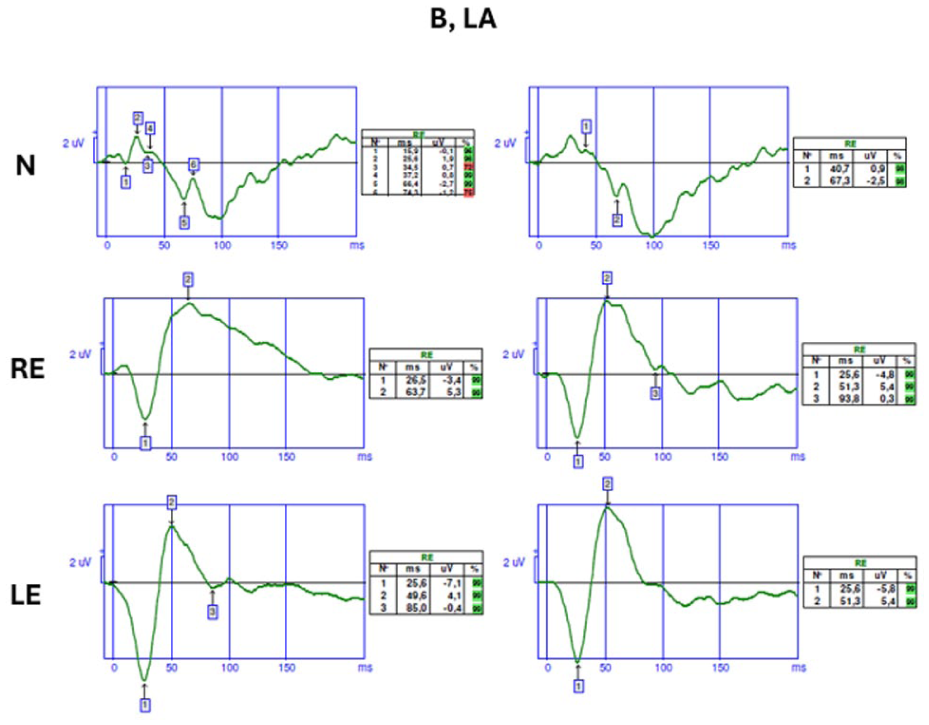
Blue-flash electroretinography responses from a healthy individual (top row) and from the patient’s right and left eyes (middle and bottom rows, respectively) demonstrating markedly increased amplitudes in both eyes.
Genetic testing revealed a homozygous mutation in the NR2E3 gene. Electrophysiologic testing demonstrated reduced rod responses, with a significant increase in blue cone responses, supporting a clinical diagnosis of enhanced S-cone syndrome. The patient was started on oral acetazolamide 250 mg thrice daily for treatment of the foveal cystic lesions. At the 2-month follow-up, BCVA improved to 20/25 OD and 20/40 OS, with a notable reduction in the foveoschisis observed on optical coherence tomography (Figure 6). Continued treatment with topical brinzolamide twice daily maintained a stable condition at the 6-month follow-up. The patient remained under monitoring at the time of writing this report.
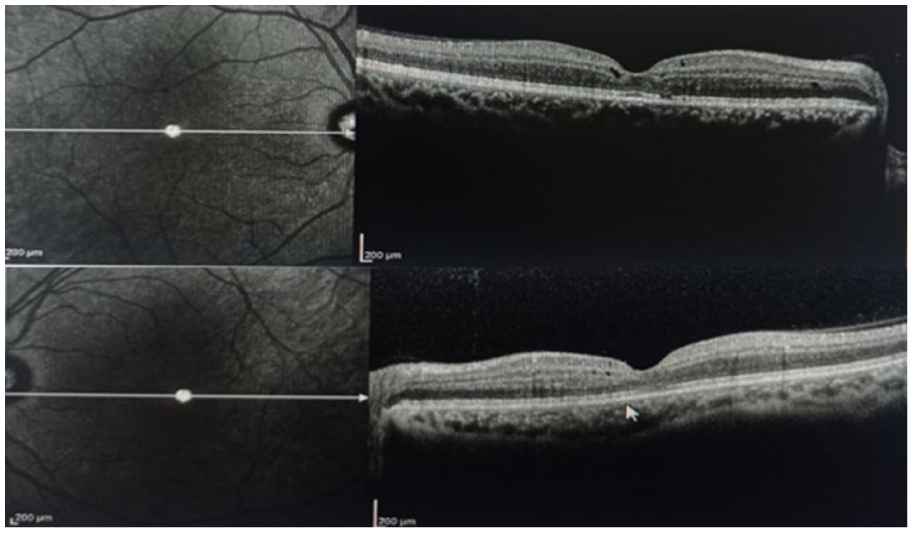
Optical coherence tomography images obtained at the 2-month follow-up demonstrating a noticeable reduction of the bilateral foveoschisis and associated intraretinal cystoid spaces after treatment.
Conclusions
The NR2E3 gene is located on chromosome 15q23 and comprises 8 coding exons. A reduction in NR2E3 expression causes photoreceptor cells to remain in their primordial S-cone form. Consequently, diseases associated with NR2E3 mutations are characterized by a reduction in rod, M-cone, and L-cone photoreceptors, along with an increase in S-cone photoreceptors. Mutations in the NR2E3 gene are known to be associated with enhanced S-cone syndrome. 2 In a postmortem analysis of a patient with enhanced S-cone syndrome harboring a homozygous NR2E3 mutation, Milam et al reported that the number of cone photoreceptors had increased to twice the normal amount, with a complete absence of rod photoreceptors. Furthermore, 92% of the cone photoreceptors were identified as S-cones. 5
Patients with enhanced S-cone syndrome usually present with nyctalopia in early childhood, which may or may not be accompanied by reduced vision. Fundus findings are highly variable, and various phenotypic manifestations have been documented. The most common fundus finding is the characteristic nummular pigmentary change distributed around the vascular arcades. In some patients, these pigmentary changes may be accompanied by white-yellow dots around the arcades as well as foveoschisis, also known as nonvasogenic cystoid maculopathy. The definitive diagnosis of enhanced S-cone syndrome is established by the presence of pathognomonic ERG findings, including reduced rod responses, delayed and reduced flicker responses, and increased short-wavelength responses, along with genetic testing.6,7
The pathophysiology of nonvasogenic cystoid maculopathy, which is observed in varying degrees in patients with enhanced S-cone syndrome, has not been fully understood. However, it is believed to be associated with the increased number of S-cones, leading to disorganization in the retinal laminar structure and associated synaptic changes. In addition to its role in photoreceptor differentiation, the NR2E3 gene is believed to maintain the integrity of the retinal structure, and mutations in this gene may therefore affect synaptic architecture. The absence of leakage on fluorescein angiography in these patients rules out the disruption of the blood–retinal barrier as the underlying mechanism of the cystoid macular changes. 8
In patients with enhanced S-cone syndrome, nonvasogenic cystoid maculopathy is a major cause of reduced visual acuity. While various treatment methods have been described, none have proved to be effective. Case reports in the literature show that topical dorzolamide usage has been associated with a reduction in cystoid changes.9,10 In a case report by Iannaccone et al, 11 oral carbonic anhydrase inhibitor therapy was administered to a patient diagnosed with enhanced S-cone syndrome, and the authors suggested that, similar to its use in X-linked retinoschisis, oral carbonic anhydrase inhibitors may also be beneficial in the treatment of enhanced S-cone syndrome. It has been hypothesized that carbonic anhydrase inhibitors influence ionic balance in the subretinal space, thereby facilitating more rapid resolution of cystoid changes associated with nonvasogenic cystoid maculopathy. 8
In our case, after 2 months of oral carbonic anhydrase inhibitor treatment, the foveoschisis almost disappeared, and the BCVA improved to 20/25 OU. This improvement was subsequently maintained with topical carbonic anhydrase inhibitor therapy.
In conclusion, although our case did not demonstrate the typical fundus findings of enhanced S-cone syndrome, the pathognomonic reduced rod responses, increased S-cone responses, and delayed and reduced flicker responses on ERG, along with identification of a homozygous NR2E3 mutation previously associated with enhanced S-cone syndrome, supported the diagnosis of an atypical form of enhanced S-cone syndrome. Most patients with enhanced S-cone syndrome present with distinct clinical profiles, highlighting the importance of individualized approaches for management, follow-up, and treatment protocols.
Consequently, identification of the underlying genetic variation is critically important, as it may aid in prognostic evaluations, guide treatment strategies, and enhance family counseling.
Footnotes
Ethical Considerations
This case report was conducted in accordance with the Declaration of Helsinki.
Consent to Participate
Informed consent was not obtained because no patient identifiers were included.
Consent for Publication
No informed consent was necessary for the publication of this paper because it does not include images or information that identifies the patient.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.