
Abstract
Introduction
Antivascular endothelial growth factor (anti-VEGF) medications have revolutionized retinal care for neovascular age-related macular degeneration (nAMD); however, this innovation has a large economic footprint. Medicare Part B claims data suggest expenditures of roughly $10,000 annually per beneficiary receiving medications such as ranibizumab and aflibercept. 1 The large cost differential between off-label bevacizumab and alternate branded agents has fueled comparative efficacy work. The landmark Comparison of Age-related Macular Degeneration Treatment Trial (CATT) showed that off-label bevacizumab is equally efficacious for nAMD, despite being significantly less costly for 2 years of treatment. 2
Cost savings from the use of lower-cost agents such as off-label bevacizumab has led to tens of billions of dollars’ worth of savings for Medicare and society. 3 Since CATT, newer biosimilar medications and branded drugs have been introduced to the nAMD landscape. 4 There is concern that US Food and Drug Administration-approved ranibizumab biosimilars, such as ranibizumab-nuna and ranibizumab-eqr, may inadvertently increase costs to the healthcare system and patients, given that these medications may be priced higher than off-label bevacizumab. 5 Therefore, exploration of the utilization and impact of biosimilars is warranted.
Given the efficacy and magnitude of savings associated with lower-cost medications, “step therapy” approaches have been designed as a form of prior authorization with which payers can mandate the use of certain lower-cost prescription medications prior to approving coverage of more expensive alternative medications. However, insurance-mandated step therapy policy is controversial for many reasons, including the prohibition of personalized medicine, reduced patient and physician autonomy, increased administrative burden, and unnecessary delays in care. 6 Medications to which patients are not responding may be continued until transition approval.
In general, across 17 of the largest US commercial health plans, 55.6% of step therapy protocols were found to be more stringent than the corresponding clinical guidelines, suggesting overly restrictive barriers to appropriate therapy initiation. 7 Prior authorization denials for anti-VEGF medications were recently found to be most commonly associated with step therapy requests. 8 While step therapy is intended to reduce the societal cost burden associated with high-cost medication, there is a lack of clarity as to the true cost savings from these approaches, especially with the addition of new biosimilar agents. Cost savings are also dependent on how often medication changes are needed for patients with refractory disease. For example, the Diabetic Retinopathy Clinical Research Network Protocol AC found no significant clinical difference between aflibercept monotherapy and bevacizumab first therapy for diabetic macular edema 9 ; however, the cost savings from this approach was more modest than expected due to the high incidence of patients switched to higher-cost medication in the clinical trial. 10
The emergence of biosimilars and agents of varying cost, durability, and clinical superiority, make step therapy’s justification and cost savings less clear. While there is evidence of these agents’ comparability with ranibizumab, 11 there is a lack of robust comparative data regarding therapeutic efficacy across the growing armamentarium for nAMD. The American Academy of Ophthalmology IRIS (Intelligent Research in Sight) Registry database provides the opportunity to assess these medication transitions among a larger cohort of patients across a wider geographic area. Consequently, we sought to characterize medication transitions of intravitreal injections for patients with nAMD in the IRIS Registry, and using real-world data, to evaluate delays in care as well as project the economic implications of medication transitions.
Methods
The IRIS Registry is a centralized data repository and reporting tool that can be used for research purposes. Research using IRIS does not constitute human subject research because data in the registry are de-identified, and the investigator does not have access to patient identifiers. Therefore, institutional board review and informed consent are not required. This study adheres to the tenets of the Declaration of Helsinki. 12 Study data are available upon reasonable request.
Inclusion and Exclusion Criteria
The IRIS Registry was frozen in July 2024 and accessed on October 20, 2024. Records spanning 2015 to July 2024 were queried for nAMD (ICD-10 codes H35.32, H35.321, H35.3210, H35.3211, H35.322, H35.3220, H35.3221, H35.323, H35.3230, H35.3231, H35.329, H35.3290, H35.3291) who received intravitreal injection (Common Procedural Terminology code 67028) any time on the day of or following nAMD diagnosis, with no documentation of intravitreal injection or medication codes prior to the index date. These criteria applied across the inclusion period and controlled for laterality. Medications were classified as bevacizumab, (HCPCS Level II drug codes J9035, C9257), biosimilar (HCPCS Level II drug codes Q5124, Q5128), and branded (HCPCS Level II drug codes J0177, J0178, J2777).
Data for patients with coexisting pathology, which would be a separate indication for intravitreal therapies, were excluded. Exclusion diagnoses include diabetic retinopathy or macular edema, branch or central retinal vein occlusion, any unspecified retinal edema, or vitreous hemorrhage in the study eye. Additionally, a history of rhegmatogenous retinal detachment or macular hole in the study eye, pars plana vitrectomy in the study eye, glaucoma, and choroidal neovascular membrane owing to other causes (eg, infections, traumatic, myopic, or inflammatory) were excluded. The study eye was defined by index injection.
Sociodemographic attributes collected include age, race and ethnicity, sex, household income in 5-digit zip code, high school graduation percentage in 5-digit zip code, insurance type, and preinjection visual acuity (VA). Zip code–level variables, median household income, and high school graduation percentage were derived from 2021 American Community Survey estimates. All available VA and central subfield thickness (CST) records in the 2 years following the first injection date were included in the longitudinal analysis.
Medication transitions were defined by the medication codes listed previously, only for patients initially treated with bevacizumab. Loss to follow-up in IRIS Registry studies for nAMD has been defined as 12 months from last intravitreal injection, with nonpersistence defined as no follow-up within 6 months from last intravitreal injection. 13 To be conservative, eyes with a gap in injections spanning 6 or more months were excluded from the present analysis. Medications were grouped as bevacizumab, biosimilar (ranibizumab nuna and eqrn), and branded (aflibercept, aflibercept HD, or faricimab). A subanalysis explored differences in branded agents. Sensitivity analysis found no differences across branded medications for interval extension; therefore, these agents were grouped in the base analysis. Given the low proportion of patients who were transitioned from a ranibizumab biosimilar to branded ranibizumab, ranibizumab was excluded from the branded group.
Cohorts were defined as bevacizumab only, bevacizumab followed by a biosimilar, bevacizumab followed by a biosimilar and then by a branded medication, and bevacizumab followed by a branded agent. The bevacizumab to biosimilar to branded group was hypothesized to represent a real-world “step therapy” cohort undergoing sequential treatment of all 3 categories of medication.
Data Analysis
Statistical analyses were performed using R (version 4.4.2) with an ɑ level (P value threshold) of .05 to determine statistical significance. Descriptive statistics compare distributions of demographic and clinical variables across the 4 treatment cohorts. Categorical variables are summarized as counts and proportions, while continuous variables are reported as medians and IQRs. Statistical comparisons included χ2 tests for categorical variables and Kruskal-Wallis tests for continuous variables.
To assess the frequency of medication transitions, we first calculated the proportion of eyes that underwent at least 1 transition. A logistic regression model was then used to evaluate the association between medication transition (binary outcome) and demographic and clinical covariates.
To assess the impact of medication transitions on delays in care, we first computed the number of days from the last injection for each eye, defining this as the dependent variable. We used a linear mixed-effects model to assess whether the interval between injections varied based on whether an eye had undergone a medication transition (coded as 1) or continued its existing medication (coded as 0). The model included a random effect for the eye and adjusted for index VA. An interaction model was also fitted to allow the effect of medication transition to vary by cohort. Given that the bevacizumab-only cohort was defined by no transitions, and our primary objective was to estimate within-eye differences, this cohort was excluded from the analysis.
To evaluate whether medication transitions predict systematic changes in injection intervals over time, we replicated these models to assess differences in the days from the last injection following the first medication transition. These models provide insight into whether medication transitions lead to prolonged gaps between injections.
To estimate the association between treatment and changes in VA and CST over time, longitudinal mixed-effects models were estimated. VA and CST were modeled as dependent variables, with the primary predictor being the drug associated with the most recent injection. Fixed effects for time (in months from first injection) were included, along with a random intercept for the eye, to account for within-eye correlation. Follow-up was included for up to 2 years from the first injection. Sensitivity analyses were performed to test the robustness of findings, including variations in follow-up duration and different model specifications for time trends.
To determine the economic impact of patterns in medication usage, transition rates from the IRIS Registry cohort, as well as injection probabilities estimated from injection rates, were used in a Monte Carlo simulation with retrospective updating. This simulation only considered the average cost of medication (cost of biosimilar, defined as the mean of the average wholesale price of ranibizumab eqrn and ranibizumab nuna [$1514.34] 14 ; branded, defined as the average wholesale price of aflibercept, aflibercept HD, or faricimab [$2745.36] 14 ; bevacizumab was priced at $65.97 per injection). Average wholesale pricing was selected for the base case given the commercial use of step approaches, as a price ceiling, and costs were tested in sensitivity analyses.
A secondary analysis was performed using Medicare’s average sale pricing, with add-on and sequestration accounting, per July 2024 rates 15 to align with cohort data (cost of biosimilar defined as the mean average sale price limit of ranibizumab eqrn and ranibizumab nuna $918.76; branded defined as the mean average sale price of aflibercept, aflibercept HD, or faricimab $2179.61; bevacizumab was priced at $65.97 per injection). A subanalysis evaluated treatment differences and costs across branded options using Medicare average sales price pricing, July 2024 rates, and compared intervals, delays in transitions, and costs between aflibercept 8 mg with faricimab (11% of injections) and aflibercept 2 mg (89% of injections). 15
Treatment pathways were defined by transitions: (1) bevacizumab followed by biosimilar, followed by a branded agent; (2) bevacizumab followed by a biosimilar; (3) bevacizumab followed by a branded agent; and (4) bevacizumab without transition. The base case model simulated 10 000 patient transitions with 100 iterations over 24 months, with state transitions determined by cumulative transition rates sourced from IRIS Registry data, distributed across cycles with Bayesian retrospective updating. Given unequal variances, a Kruskal-Wallis test, followed by a Dunn’s test with Bonferroni correction, was applied using R package. Sensitivity analyses included transition rates, medication costs, and injection probabilities (increased or decreased by 10%). Additional sensitivity for time horizon beyond 2 years included 5-year and 10-year projections with 5% annual discounting and 5% inflation.
Results
A total of 182 757 eyes met inclusion criteria; cohorts were defined as bevacizumab only (136 451 eyes); bevacizumab followed by a biosimilar (2184 eyes); bevacizumab followed by a biosimilar, followed by branded medication (335 eyes); and bevacizumab followed by a branded agent (43 787 eyes). Overall, 24.56% of eyes transitioned medications during the study period.
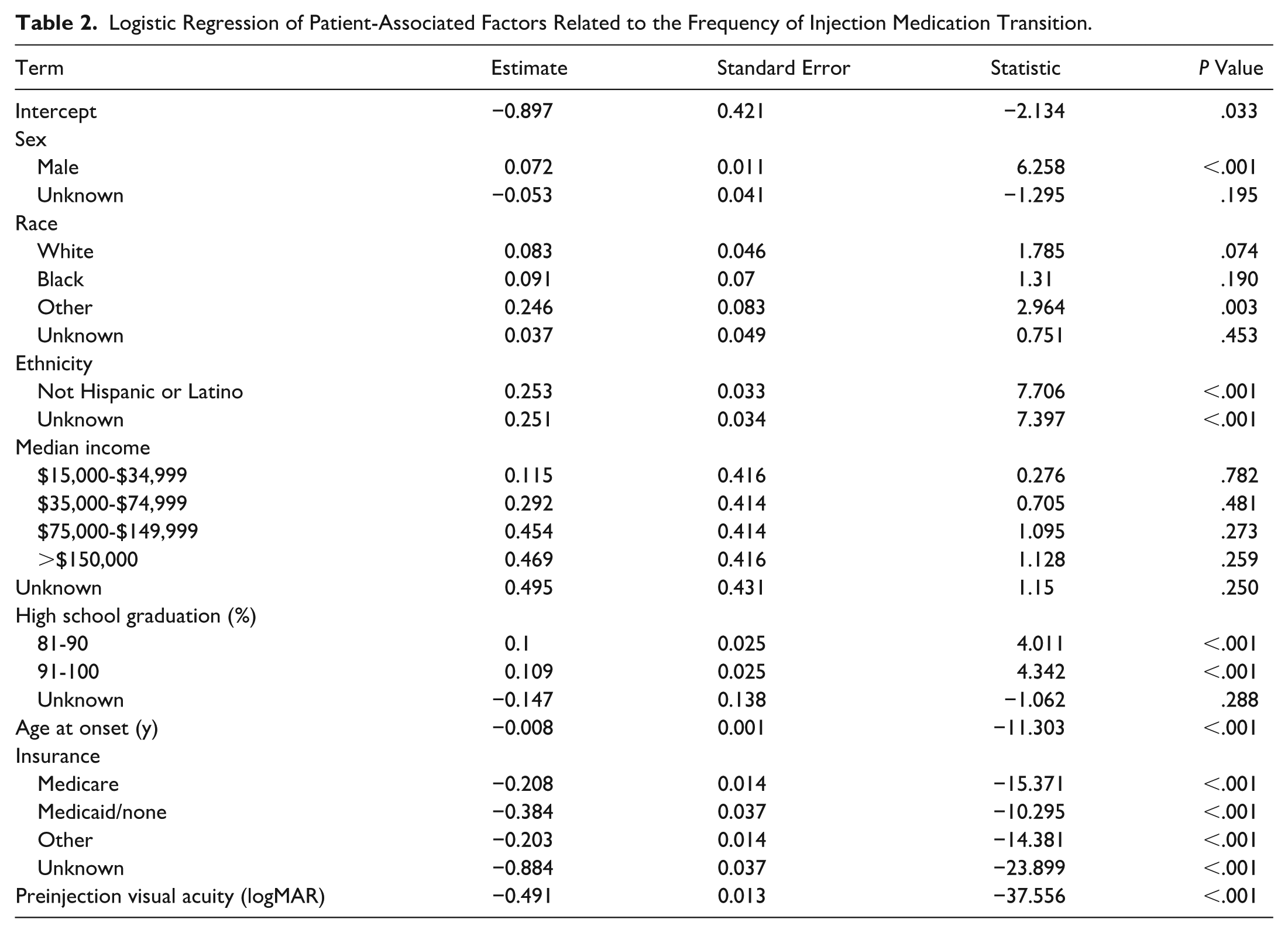
The bevacizumab-only cohort had the lowest percentage of patients with commercial insurance (28%) compared with other cohorts (bevacizumab followed by a biosimilar, 34%; bevacizumab followed by a biosimilar, followed by branded medication, 37%; and bevacizumab followed by a branded agent, 34%) and the highest percentage of patients on Medicaid (3.0%) compared with other cohorts (bevacizumab followed by a biosimilar, 2.4%; bevacizumab followed by a biosimilar, followed by branded medication, 1.2%; and bevacizumab followed by a branded agent, 2.2%) (Table 1). Patients with Medicaid and Medicare were less likely to undergo medication transition compared with commercial insurance patients (log-odds of −0.384 and −0.208, respectively; P < .001) (Table 2).
Demographic Characteristics. a
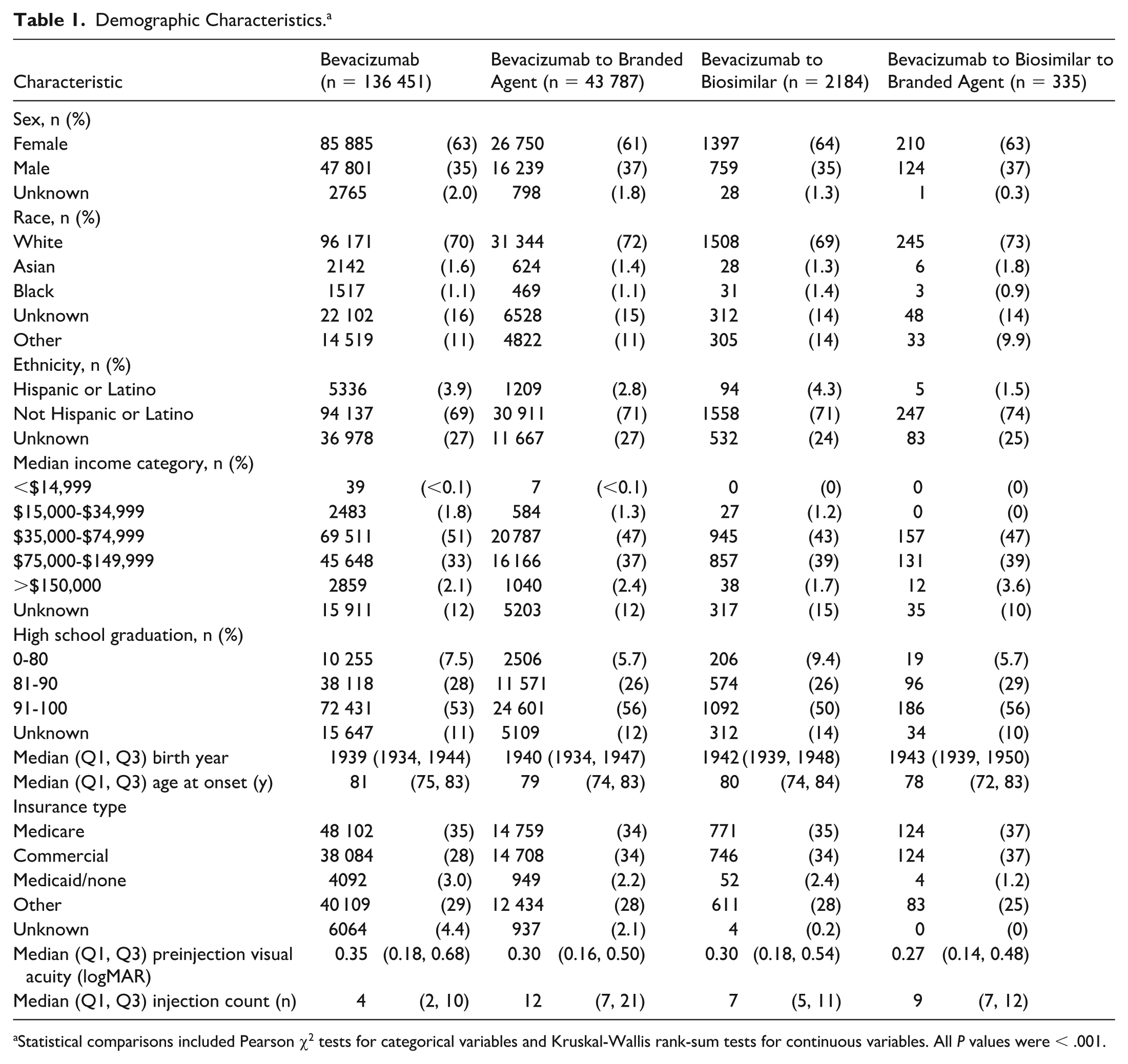
Statistical comparisons included Pearson χ2 tests for categorical variables and Kruskal-Wallis rank-sum tests for continuous variables. All P values were < .001.
Logistic Regression of Patient-Associated Factors Related to the Frequency of Injection Medication Transition.
To assess delays in care during transition periods, differences in days from the last injection were assessed across cohorts. Prior to transition, the group on bevacizumab followed by a biosimilar received injections on average every 51.4 days. After the medication transition from bevacizumab to a biosimilar, bevacizumab to a biosimilar followed by a branded agent, and bevacizumab to a branded agent, there was an additional delay of 9.849, 6.192, and 0.982 days, respectively (all P < .001), between injections compared with the immediately preceding injection interval (P < .001). Injection intervals were also assessed, with post-transition intervals showing extended time between injections. After transition, injection intervals increased by 8.465 (P < .001), 4.686 (P < .001), and 9.523 (P = .003) days for bevacizumab to a biosimilar, bevacizumab to a biosimilar followed by a branded agent, and bevacizumab to a branded agent, respectively.
The baseline cohort resulted in a higher mean cost per patient for bevacizumab followed by a biosimilar followed by branded medication, compared with bevacizumab followed by branded medication, bevacizumab followed by a biosimilar, and bevacizumab alone. In the base case cost model, all cohorts were statistically significantly different in terms of medication costs (Table 3). A Kruskal-Wallis test followed by Dunn’s test with Bonferroni correction showed bevacizumab followed by a biosimilar was 36.3% less costly than bevacizumab followed by a biosimilar followed by branded medication (P < .001), and bevacizumab followed by a branded agent was 11.62% less costly than bevacizumab followed by a biosimilar followed by branded agent (P < .001).
Baseline 2-Year Economic Simulation. a
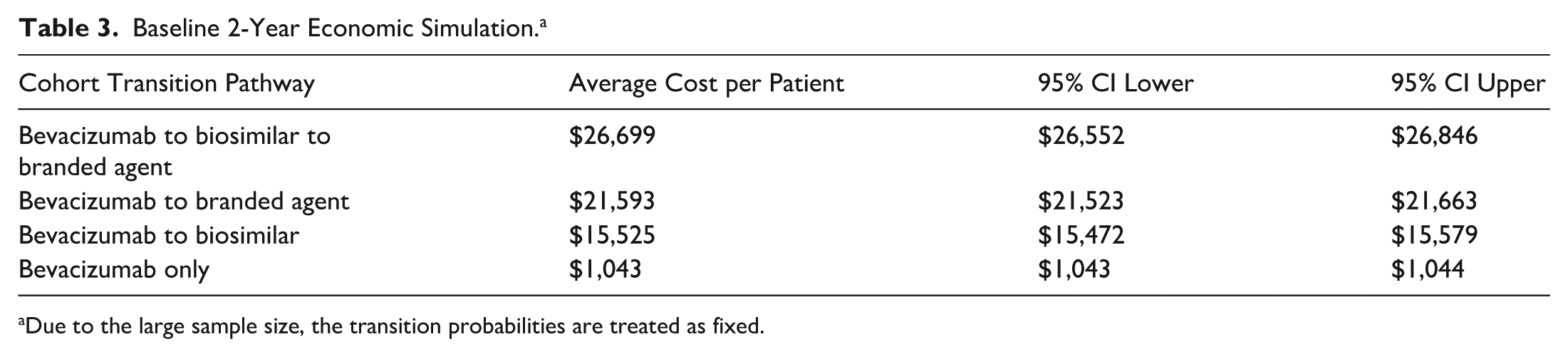
Due to the large sample size, the transition probabilities are treated as fixed.
These results were sensitive to injection frequency, cost per injection, and estimated transition probability, but the trend toward higher-cost bevacizumab followed by a biosimilar followed by branded agent persisted (Supplemental Table 1). Secondary analysis using Medicare average sale prices showed similar results, with bevacizumab followed by a biosimilar being 49.4% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001), but bevacizumab followed by a branded medication was only 1.6% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P = 1) (Supplemental Table 2). Projections for 5- and 10-year windows persisted over time for both analyses.
Subanalysis of the bevacizumab-to-branded cohort showed that there was an additional statistically significant delay between transitions for newer branded agents faricimab and aflibercept 8 mg of 8.614 days, compared with aflibercept 2 mg (P < .001); however, there was no statistically significant difference in interval extension for longer-acting agents faricimab and aflibercept 8 mg (9.79 days), compared with aflibercept 2 mg (9.46 days) (P = .118). Subanalysis across agents showed that for aflibercept 2 mg, bevacizumab followed by a biosimilar was 28.14% less costly than bevacizumab followed by a biosimilar followed by a branded medication (P < .001), and bevacizumab followed by a branded medication was 47.73% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001).
For faricimab, bevacizumab followed by a biosimilar was 58.4% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001), and bevacizumab followed by a branded medication was 27.81% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001). For aflibercept 8 mg, bevacizumab followed by a biosimilar was 65.43% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001), and bevacizumab followed by a branded medication was 23.64% less costly than bevacizumab followed by a biosimilar followed by a branded agent (P < .001). Therefore, bevacizumab followed by a biosimilar followed by a branded agent was the most costly approach across all variations. For aflibercept 2 mg, bevacizumab followed by a branded medication was 37.5% less costly than bevacizumab followed by a biosimilar (P < .001); however, for faricimab and aflibercept 8 mg, bevacizumab followed by a biosimilar was 42.4% (P < .001) and 54.73% (P < .001) more costly, respectively, than bevacizumab followed by a branded agent.
VA measures showed improvement in all cohorts, with a peak at around 5 months from the first injection, followed by a slow decline in VA. The differences between VA under different medications have uncertain clinical significance. CST decreased under all medications, with eyes that transitioned to a biosimilar showing a CST 2.46 μm lower than with bevacizumab (P = .090) and eyes that transitioned to branded medications showing a CST 10.88 μm lower than with bevacizumab (P ≤ .001).
Conclusions
The present analysis characterized patients receiving biosimilar intravitreal agents for nAMD within the IRIS Registry cohort, with a focused subanalysis exploring the impact of step therapy approaches for cohorts of transitioned patients. Patients who transitioned to a biosimilar experienced relatively increased delays in care during the transition period. The step therapy cohort (bevacizumab followed by a biosimilar followed by a branded agent) also demonstrated the highest frequency of injections per month, and the shortest interval extension after transition. As a result, the step therapy cohort (bevacizumab followed by a biosimilar followed by a branded agent) was more costly than direct transitions from bevacizumab to biosimilar or branded medications in economic modeling. Given minimal clinically significant differences in VA or CST, these findings suggest step therapy approaches may be associated with delays in care, less interval extension, and higher costs that deserve further research.
In particular, longer delays in care with step therapy approaches are concerning. Prior work studying prior authorization delays in retina care found 59.6% of prior authorizations resulted in delay in care greater than 24 hours, with 26.3% approved within 8 to 31 days and 12.4% approved within more than 31 days. 8 This aligns with our findings of delays in care for the medication transition from bevacizumab to a biosimilar, bevacizumab followed by a biosimilar followed by a branded agent, and bevacizumab to a branded agent of 9.849, 6.192, 0.982 days, respectively. Delays in care may lead to irreversible changes in VA, and it is possible that the longer delays from the transitions to biosimilars are due to denials for attempts to use branded alternatives.
Delays in care affecting patient outcomes are difficult to quantify, given the lack of publicly available data on denials of care, but there is growing concern that prior authorization and utilization management can create barriers to high-value treatments, promote anxiety and mistrust in the healthcare system, and contribute to adverse outcomes.16,17 While the analysis in this study was performed as stringently as possible with within-person comparisons, the delays are relative and may not represent the absolute delays in care, which would be best validated in a prospective study. In addition, the clinical significance of delays in care should be further studied.
Baseline economic modeling found that bevacizumab followed by a biosimilar followed by a branded agent is 11% higher cost than bevacizumab followed by branded agents and 36.3% higher cost than bevacizumab followed by a biosimilar, which implies the cost savings associated with blanket step therapy mandates may be uncertain. While bevacizumab only groups are expectedly lower cost, and bevacizumab patients transitioned to biosimilar terminally are lower cost, the cohort with multiple step transitions across categories had the highest frequency of injections and consequently the highest cost profile. Subanalysis showed that the bevacizumab transition to aflibercept 2 mg is less costly than the bevacizumab transition to biosimilar, whereas the bevacizumab transition to either aflibercept 8 mg or faricimab is more costly than the bevacizumab transition to biosimilar. However, aflibercept 2 mg, aflibercept 8 mg, and faricimab direct transitions from bevacizumab were each less costly than the bevacizumab followed by a biosimilar followed by a branded agent multistep pathway, suggesting that the full step therapy pathway may be the most costly approach for patients ultimately requiring branded agents.
There are known limitations in the IRIS Registry cohort, 18 including data duplication, which is relevant to injection frequency calculations performed here. This artifact may increase the number of procedures in each cohort19,20; however, this bias is expected to be uniform across cohorts. Thus, the relative comparisons presented here are methodologically valid and likely clinically meaningful. 20 The higher rate of injections and lower interval extension in patients receiving biosimilars compared with branded medications may suggest that these medications have less durability, or it may be related to practice patterns in patients requiring step therapy. Paradoxically, step therapy cohorts are possibly the costliest because of reloading practice patterns. For example, patients may be treated with frequent loading doses of biosimilar intermediaries to meet a minimum payer definition of biosimilar failure and then be transitioned to a payer-allowed branded alternative as expeditiously as possible.
There is no consensus on the injection interval for patients after medication transition, with some ophthalmologists choosing to repeat loading injections monthly, similar to clinical trials, compared with immediate attempts at treatment extension. The former hypothesis is well supported by this IRIS Registry data analysis, with the cohort receiving bevacizumab followed by a biosimilar followed by a branded agent experiencing the shortest interval extension between transitions, the most frequent injections, and subsequently, the highest modeled costs. Importantly, this analysis was unable to distinguish insurance subtypes such as Medicare Advantage plans, which may have different prior authorization requirements that could contribute to delays in care.
VA findings here show an early improvement that declines in subsequent months to years. This aligns with prior work showing VA improvement at the end of year 1, with subsequent declines despite treatment and other clinical trial data showing a decline in long-term vision in patients with nAMD, regardless of early VA gains.12,21,22 While the IRIS registry does not allow an extensive analysis of VA or optical coherence tomography findings, patients receiving biosimilar and branded injections showed lower CST than patients receiving bevacizumab injections, suggesting some additional drying benefit for refractory fluid.
While the bevacizumab-only cohort had substantial cost savings, similar to prior work,23,24 the potentially improved clinical efficacy of the branded and biosimilar cohorts, especially for bevacizumab nonresponders, requires further research. The cost results alone should not be used to advocate for longer mandates for bevacizumab-only regimens, which could inadvertently harm those bevacizumab nonresponders or incomplete responders. Further prospective, comparative multicenter studies are needed to assess clinical effectiveness.
Finally, to our knowledge, this is the first IRIS Registry study to incorporate sociodemographic covariates such as income category and high school graduation. While the demographic distribution varied minimally across cohorts, there were notable differences across insurance groups. Specifically, the bevacizumab-only cohort contained the highest percentage of patients on Medicaid and the lowest percentage of those on commercial insurance. Additionally, Medicaid and Medicare insurance were associated with a decreased likelihood of medication transition. These findings suggest there could be payer or socioeconomic factors contributing to medication access or selection, which is deserving of further study.
There are important additional limitations to the present work. Transition rates within the study window may be influenced by the Food and Drug Administration approval of aflibercept in 2011, 25 ranibizumab-nuna in 2021, 26 ranibizumab-eqrn in 2022, 27 faricimab in 2022, 28 and aflibercept HD in 2023. 29 The IRIS Registry data here represent early postapproval inclusion of biosimilars. This may explain the smaller cohort receiving bevacizumab followed by a biosimilar followed by a branded agent, and longer-term postmarket data would be helpful to determine the total cost profile of each cohort.
The current analysis focuses on a real-world cohort selectively biased by consistent follow-up. Prior IRIS Registry analysis of nAMD showed that 1 in 9 patients were lost to follow-up, with 1 in 7 defined as nonpersistent (no follow-up within 6 months from the last intravitreal injection). 13 Additional IRIS Registry analysis suggests frequent discontinuation of treatment, with more than one-third of eyes discontinuing treatment by year 3, 21 as well as a low rate of transition back to the original agent (11.8%). 12
The economic model window and approach here align with these findings but are limited by study timeframe, specific transition pathways, and pricing assumptions. These were tested in scenario analysis for inflationary adjustments, discounting, and Medicare average sale price pricing. These scenarios found similar results but less robust differences in pathways. Further study is needed to test these limitations, but the evidence from this real-world cohort suggests current biosimilar usage and economic impacts may not inherently be cost-effective.
The present analysis found concerning differences in nAMD medication transitions. In addition to demographic differences in medication transitions, biosimilar use was associated with delays in care, and economic modeling suggests that multiple transitions may not be cost-effective. Given the current use of step therapy mandates and the potential for irreversible vision loss with delays in care, the findings here suggest that multiple transitions can inadvertently promote higher-cost, lower-value approaches than physician-directed care. The true societal or payer perspective of cost savings will depend on the proportion of patients ultimately requiring multiple transitions, which cannot be determined from this cohort.
Regardless, these findings suggest that payer-related policy revisions may be necessary to provide the highest-value care to patients. Future work on the durability of new agents should explore the cost utility of branded-first approaches, which could provide advantageous economic and clinical results in certain populations.
Supplemental Material
sj-docx-1-vrd-10.1177_24741264261455206 – Supplemental material for Intravitreal Anti-VEGF Injection Transitions Across Bevacizumab, Biosimilar, and Branded Medications: The IRIS Registry Study
Supplemental material, sj-docx-1-vrd-10.1177_24741264261455206 for Intravitreal Anti-VEGF Injection Transitions Across Bevacizumab, Biosimilar, and Branded Medications: The IRIS Registry Study by Sean T. Berkowitz, Joshua B. Gilbert, Connor J. Ross, William C. Kearney, Alice Lorch, Joan W. Miller, Elizabeth J. Rossin, Paul Sternberg and Avni P. Finn in Journal of VitreoRetinal Diseases
Supplemental Material
sj-docx-2-vrd-10.1177_24741264261455206 – Supplemental material for Intravitreal Anti-VEGF Injection Transitions Across Bevacizumab, Biosimilar, and Branded Medications: The IRIS Registry Study
Supplemental material, sj-docx-2-vrd-10.1177_24741264261455206 for Intravitreal Anti-VEGF Injection Transitions Across Bevacizumab, Biosimilar, and Branded Medications: The IRIS Registry Study by Sean T. Berkowitz, Joshua B. Gilbert, Connor J. Ross, William C. Kearney, Alice Lorch, Joan W. Miller, Elizabeth J. Rossin, Paul Sternberg and Avni P. Finn in Journal of VitreoRetinal Diseases
Footnotes
Author Note
J.B. Gilbert provided statistical expertise for this study.
Ethical Considerations
This study adheres to the tenets of the Declaration of Helsinki.
Consent to Participate
Informed consent and patient consent are not applicable to this study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported in part by a Research to Prevent Blindness unrestricted grant to the Vanderbilt Eye Institute. The sponsor or funding organization had no role in the design or conduct of this research
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Miller receives royalties from Bausch + Lomb, Mass Eye and Ear, and QLT Inc.; and is a consultant for ONL Therapeutics, LLC, and Sumitomo Pharma America, Inc. Dr. Rossin is a consultant for AbbVie. Dr. Sternberg is a consultant for the Association of University Professors of Ophthalmology; and is on the board of the International Retina Research Foundation, the National Alliance for Eye and Vision Research/Alliance for Eye and Vision Research, and the Usher’s III Initiative. Dr. Finn is on the advisory board of AbbVie, Alimera, Apellis, and Iveric Bio; is a consultant for Bausch + Lomb and Genentech; and does research for EyePoint. None of the other authors declared potential conflicts of interest with respect to the research, authorship, and/or publication of the article.
Supplemental Material
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.