
Abstract
Introduction
Papillorenal syndrome, also known as renal-coloboma syndrome, is a rare congenital autosomal dominant inherited condition characterized by variable phenotypic manifestations affecting ocular and kidney development. Approximately half of the affected patients harbor a mutation in the paired box gene PAX2 located on chromosome 10q24. 1 Ocular findings are variable and range from normal vision with mild structural changes to significant optic nerve excavation and severe vision loss associated with maculopathy or retinal detachment.2–4 The prevalence of maculopathy and retinal detachment in papillorenal syndrome remains unknown due to the rarity of this condition. The management of associated maculopathy is variable, with no clear consensus in the literature. This variability is partly attributable to the variable optic nerve findings and inconsistent nomenclature used to describe these lesions, including optic nerve coloboma, morning glory disc anomaly, optic nerve excavation, and optic nerve pit.2–5 While some authors advocate observation or medical management, others have reported success with pars plana vitrectomy (PPV) and laser.3,6–8
Herein, we describe 3 eyes from 2 patients with papillorenal syndrome-associated maculopathy. Two of the eyes were treated with PPV, endolaser to the temporal peripapillary retina, and tamponade, and 1 eye was treated with indirect laser photocoagulation alone. These cases demonstrate that untreated maculopathy may lead to progressive vision loss; however, the exact timing at which intervention should be recommended remains uncertain.
Case Reports
Case 1
A 12-year-old boy with no significant past medical history presented with decreased vision in the left eye. On examination, visual acuity (VA) was 20/20 OD and 20/400 OS. Intraocular pressure was 15 mm Hg OD and 12 mm Hg OS. Anterior segment examination showed no abnormalities. Slitlamp examination showed an anomalous optic nerve with tortuous vessels and temporal rim thinning in both eyes. The macula appeared mildly thickened in the right eye, whereas subretinal fluid (SRF) was observed in the left eye (Figure 1). Peripheral retinal examination was unremarkable.
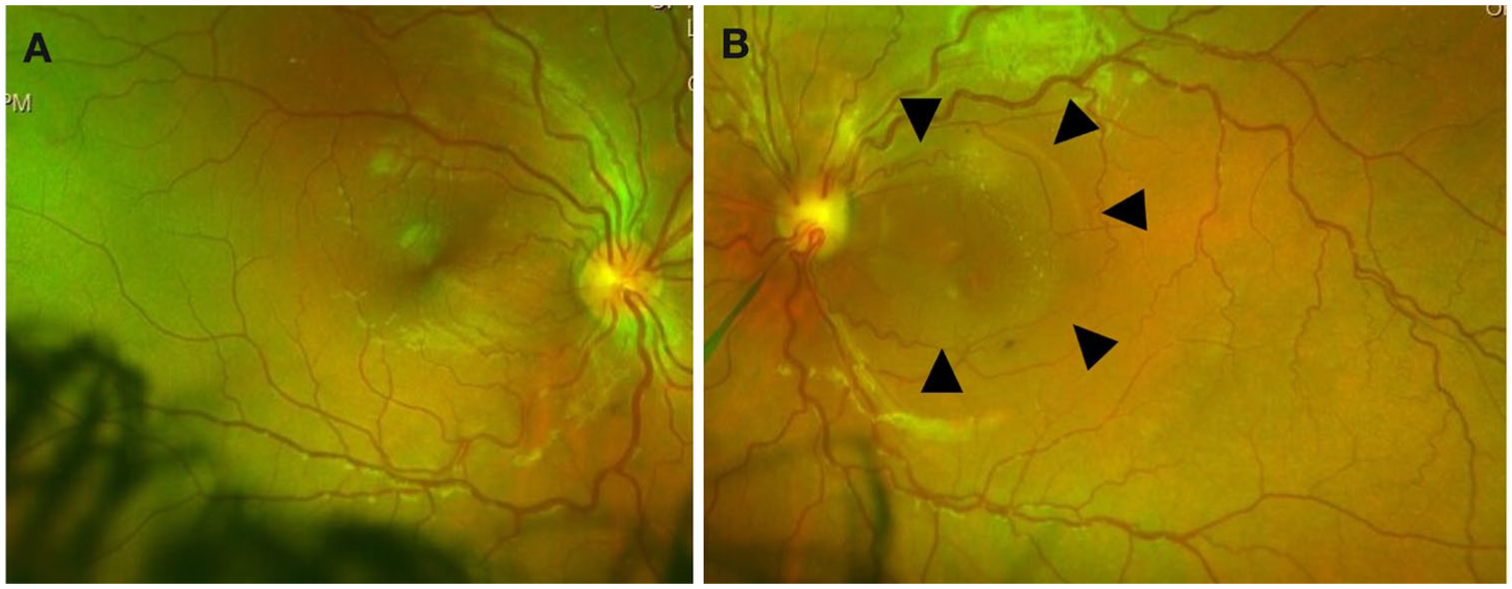
Color fundus photographs of the (A) right and (B) left eyes demonstrating anomalous optic nerves with tortuous retinal vessels, increased vascular density, and temporal rim thinning. (B) In the left eye, arrowheads indicate changes in the macular reflex attributed to the presence of intraretinal and subretinal fluid.
Optical coherence tomography (OCT) confirmed the presence of intraretinal fluid (IRF) in the right eye and both SRF and IRF in the left eye (Figure 2). After no improvement and mild worsening of the fluid 3 months after initial presentation, surgical treatment was recommended for the left eye. The patient underwent 23-gauge PPV with light laser photocoagulation to the superotemporal quadrant of the peripapillary retina and silicone oil tamponade. Induction of a posterior vitreous detachment (PVD) was not possible during the initial surgery.
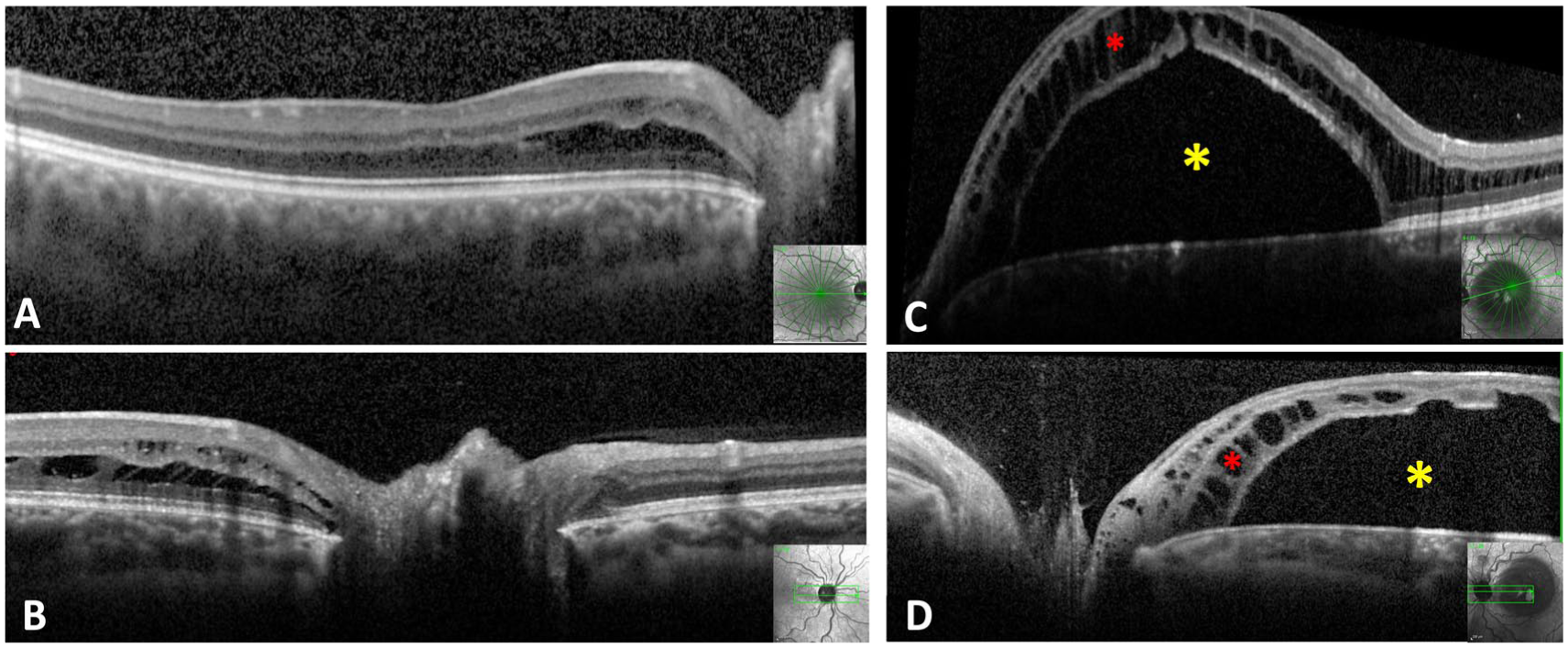
Optical coherence tomography (OCT) findings at presentation. Right eye: (A) Macular OCT demonstrating intraretinal fluid (IRF) and (B) peripapillary OCT showing schisis-like IRF. Left eye: (C) Macular OCT and (D) peripapillary OCT demonstrating significant subretinal fluid (yellow asterisks) associated with schisis-like IRF (red asterisks).
At the 1-year follow-up, OCT demonstrated initial mild improvement of the SRF and IRF in the left eye; however, this improvement plateaued without complete resolution after the first surgery. A second surgery was recommended and consisted of silicone oil removal, complete induction of a PVD, additional light laser photocoagulation to the temporal peripapillary retina, and 14% perfluoropropane gas tamponade with postoperative face-down positioning. Complete resolution of the fluid in the left eye was achieved 15 months after the second intervention (Figure 3). Best-corrected VA (BCVA) was 20/20 OD and 20/400 OS. During follow-up, the right eye demonstrated worsening IRF with a mild decrease in VA (Figure 3). Light laser photocoagulation to the temporal peripapillary retina was subsequently performed under general anesthesia using an indirect ophthalmoscope. The IRF completely resolved 2 years after treatment.
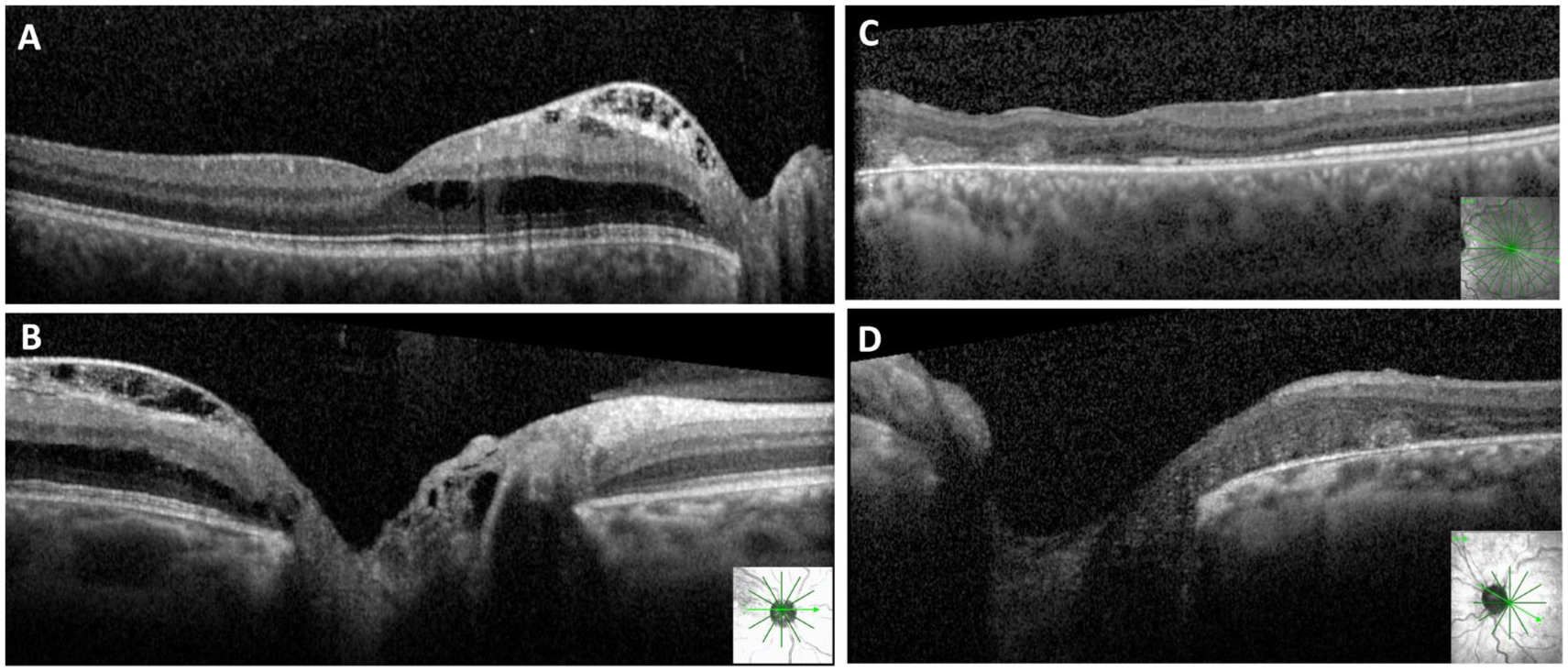
Optical coherence tomography (OCT) findings during follow-up. Right eye: (A) Macular OCT and (B) peripapillary OCT demonstrating worsening intraretinal fluid (IRF). Left eye: (C) Macular OCT and (D) peripapillary OCT demonstrating complete resolution of IRF and subretinal fluid 15 months after surgery.
At the final follow-up examination 9 years after the initial presentation, BCVA was 20/20 OD and 20/200 OS. Fundus examination demonstrated bilateral optic nerve rim thinning, which was more pronounced in the left eye (Figure 4). The morphology of the optic nerves more clearly demonstrated retinal vessels emanating from the rim of the optic nerve, a feature characteristic of papillorenal syndrome. Genetic testing identified a pathogenic PAX2 mutation, c.685C>T (p.Arg229*). OCT demonstrated complete resolution of the IRF and SRF in both eyes (Figure 5). Loss of photoreceptors was present in the left eye, which likely accounted for the limited final visual outcome.
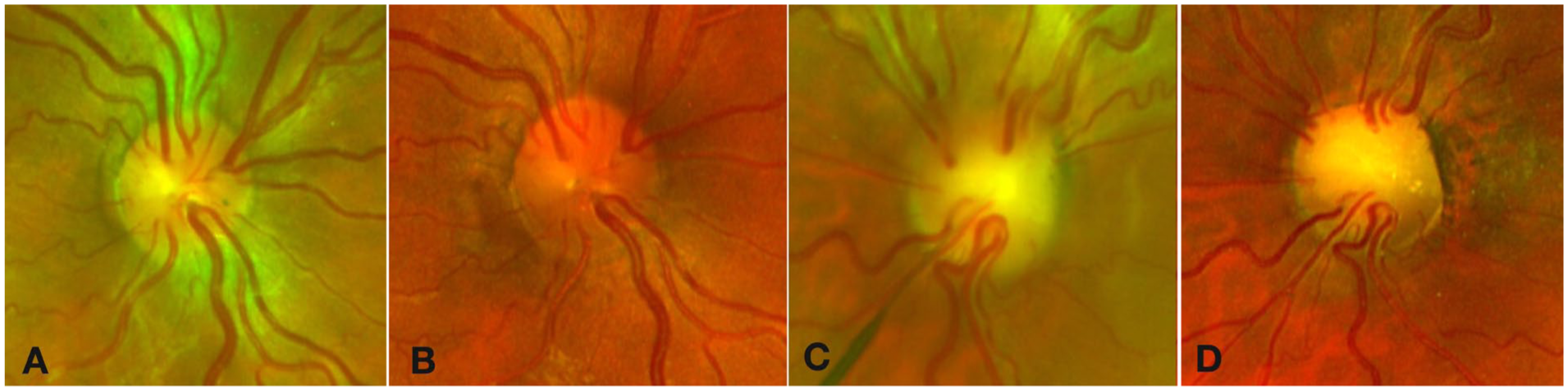
Serial color fundus photographs demonstrating optic nerve progression over 9 years of follow-up. Right eye: (A) At presentation and (B) at 9-year follow-up. Left eye: (C) At presentation and (D) at 9-year follow-up. Both eyes demonstrate progressive thinning of the optic nerve rim.
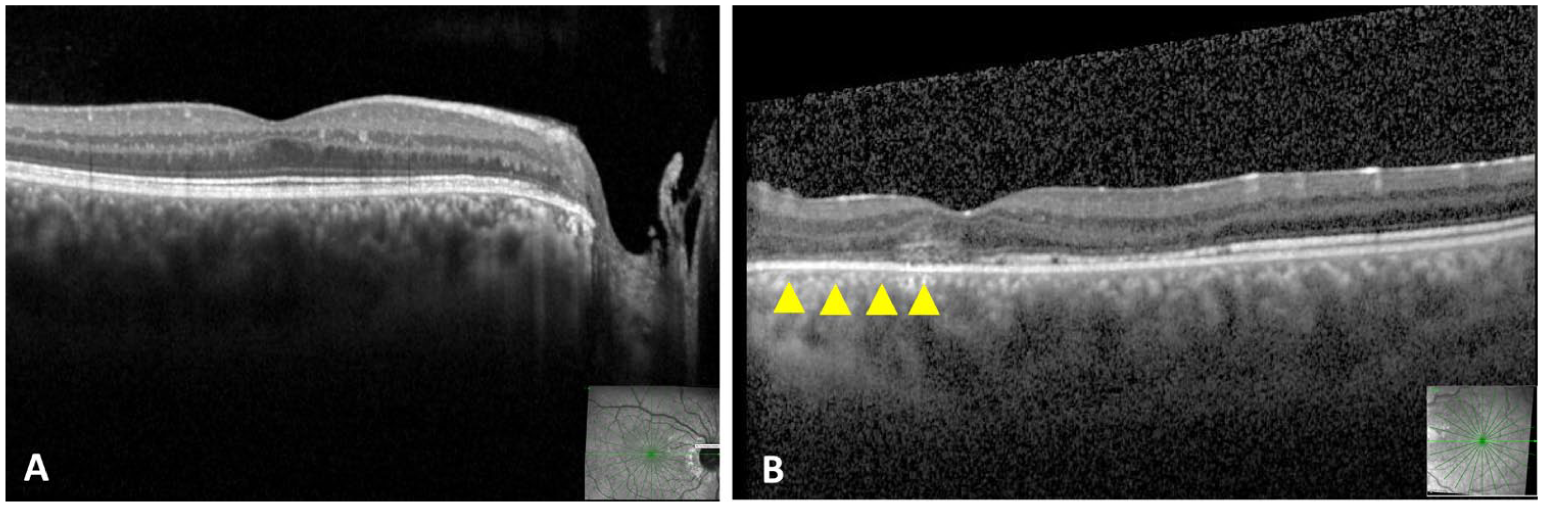
Optical coherence tomography (OCT) images at final follow-up. Right eye: (A) OCT through the optic nerve and macula demonstrating complete resolution of the intraretinal fluid (IRF) after laser treatment. Left eye: (B) Macular OCT demonstrating resolution of both subretinal fluid and IRF after surgical intervention. Yellow arrowheads indicate the loss and attenuation of the photoreceptor layer in the left eye.
Further review of the family history revealed that the patient’s mother was awaiting kidney transplantation due to a long-standing history of kidney disease of unknown etiology despite multiple biopsies. Genetic testing identified the same PAX2 mutation in the mother. Nephrology consultation for the patient, as well as ophthalmologic examination of his mother and siblings, was recommended.
Case 2
An 11-year-old boy was referred by a pediatric ophthalmologist for evaluation of decreased vision in the left eye, previously diagnosed as “morning glory disc anomaly associated with macular edema.” Previously performed brain magnetic resonance imaging and magnetic resonance angiography were reported as unremarkable. At presentation, BCVA was 20/40 OD and 20/50 OS. Anterior segment examination showed no abnormalities. Slitlamp examination showed anomalous optic nerves in both eyes, with tortuous blood vessels emanating from a thin rim (Figure 6). The macula appeared thickened in the right eye and flat in the left eye. Peripheral retinal examination was grossly unremarkable. OCT confirmed the presence of IRF in the right eye, with no IFR or SRF in the left eye (Figure 7).
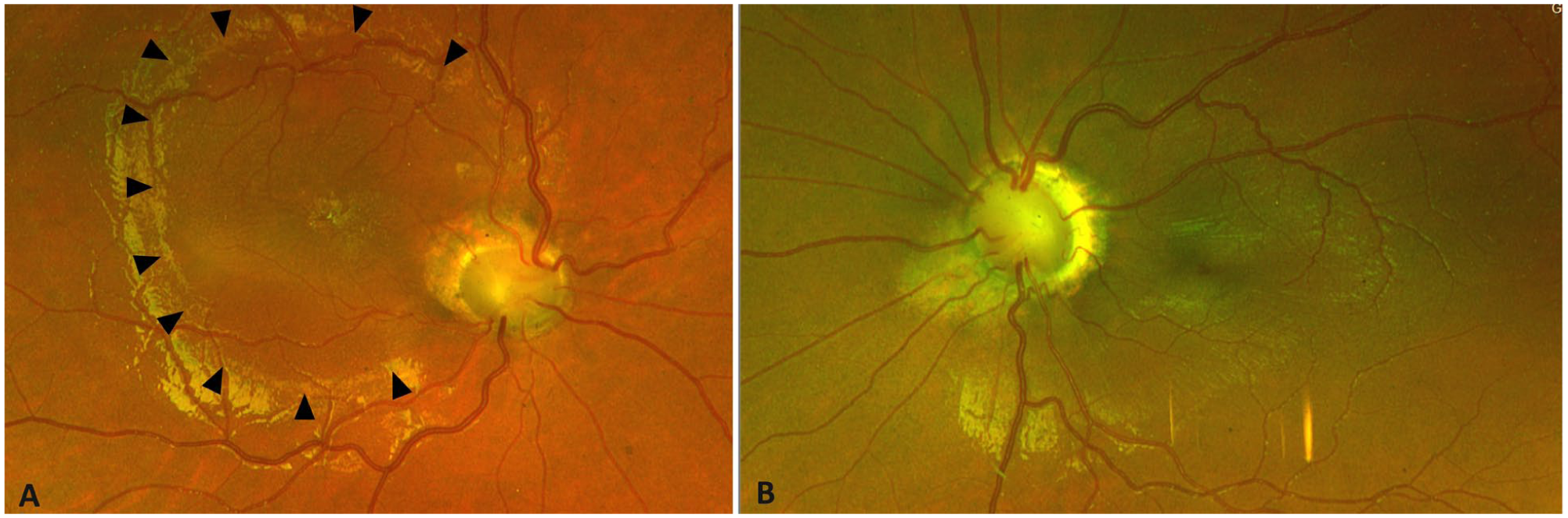
Color fundus photographs of Case 2. Right eye: (A) Anomalous optic nerve with tortuous blood vessels emanating from a thin rim and associated macular thickening. Arrowheads indicate changes in the macular reflex attributed to the presence of intraretinal fluid. Left eye: (B) Anomalous optic nerve with tortuous retinal vessels emanating from a thin neuroretinal rim. The macula appears flat.
Optical coherence tomography (OCT) images of Case 2 at presentation. Right eye: (A) OCT demonstrating an excavated optic disc associated with schisis-like intraretinal fluid (IRF) extending into the macula in a straight columnar pattern. Left eye: (B) OCT demonstrating an excavated optic disc with no IRF or subretinal fluid but mild outer retinal changes.
Given the appearance of the optic nerves, genetic testing and close observation were recommended. Follow-up genetic testing identified a pathogenic PAX2 mutation, c.310C>T (p.Arg104*). Further review of the patient’s past medical history revealed that the patient was diagnosed with renal hypoplasia and was awaiting kidney transplantation. At that time, VA and the volume of IRF in both eyes remained stable. Examination under anesthesia (EUA) with possible treatment to the right eye was recommended; however, the patient’s mother declined due to his ongoing kidney problems and possible need for kidney surgery.
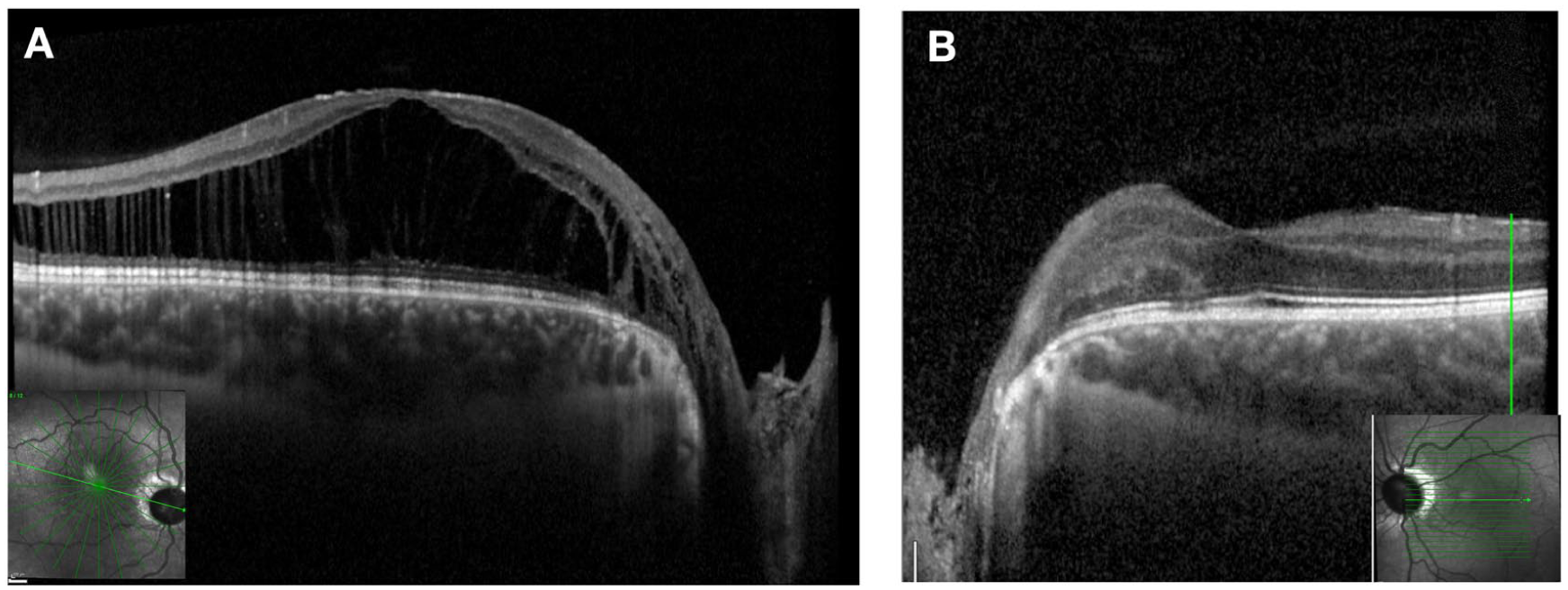
The patient returned 10 months later after seeking a second opinion and undergoing treatment with topical dorzolamide, ketorolac, and prednisolone. During this period, his condition worsened, and he developed increased intraocular pressure due to topical corticosteroid use. On examination, BCVA was 20/100 OD and 20/50 OS. OCT demonstrated worsening IRF and new SRF in the right eye, while no change was observed in the left eye (Figure 8). At that time, EUA and surgical intervention were recommended. The patient underwent 25-gauge PPV with PVD induction, light laser photocoagulation to the temporal peripapillary retina, and 12% perfluoropropane (C3F8) gas tamponade, with strict instructions to maintain postoperative face-down positioning for the first 2 weeks. At the most recent follow-up, 10 months after surgery, OCT demonstrated significant improvement in both IRF and SRF, with BCVA improving to 20/40. The SRF had resolved under the fovea and continued to decrease elsewhere (Figure 9).
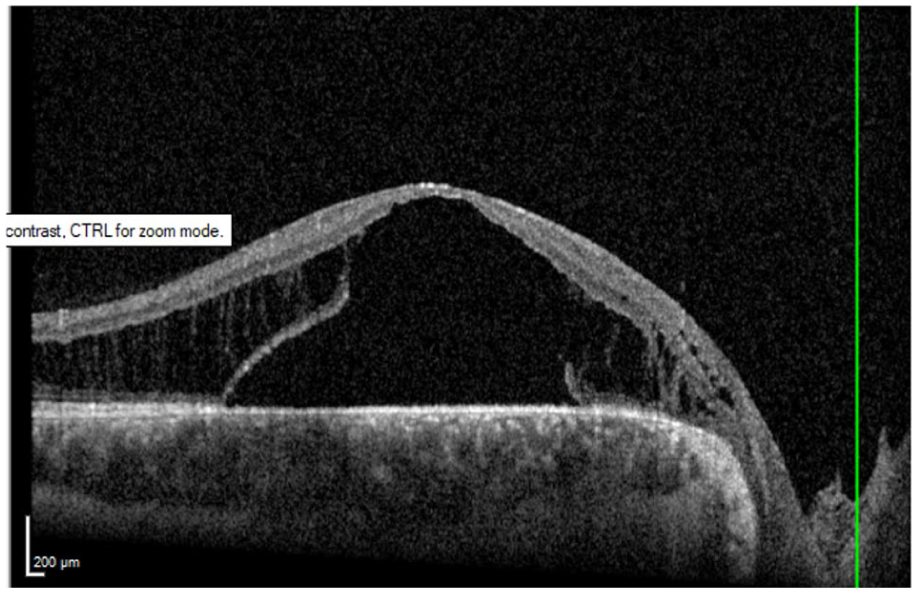
Optical coherence tomography image of the right eye demonstrating the presence of subretinal fluid with thickening of the macula after treatment with topical dorzolamide, prednisolone, and ketorolac.
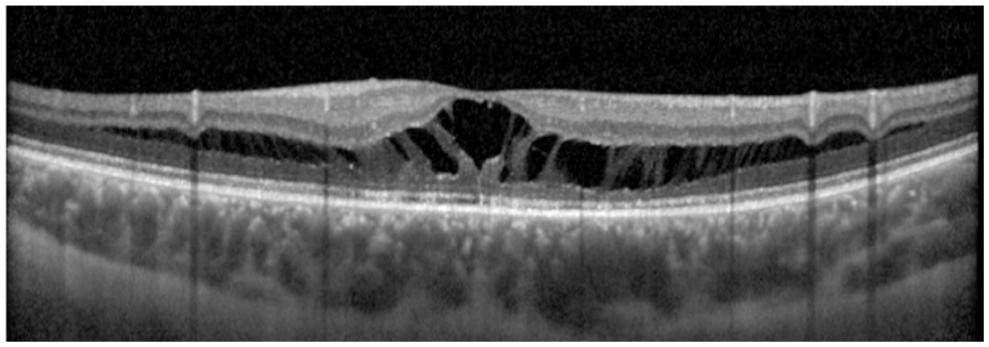
Optical coherence tomography image of the right eye after surgery showing marked improvement of the maculopathy. Residual schisis-like intraretinal fluid is present, with complete resolution of subfoveal fluid and continued reduction in fluid elsewhere at the most recent follow-up 10 months after surgery.
Conclusions
Reis first described optic disc pit and its associated maculopathy, or serous macular detachment, in 1908. 9 Since then, various mechanisms have been proposed to explain these findings, leading to a diverse array of treatment strategies. Similar forms of maculopathy and serous retinal detachment have also been observed in association with other optic nerve anomalies, such as staphyloma, morning glory disc anomaly, and optic nerve coloboma. Until the underlying causes are more clearly understood, the term “optic pit-like maculopathy” may serve as the most straightforward term for these entities.
Given the rarity of papillorenal syndrome and its variable phenotypic manifestations, management of the associated maculopathy remains inconsistent and has historically ranged from observation and medical therapy to surgical intervention.2–8 One reason why management strategies remain poorly determined is the lack of uniform nomenclature used to describe the optic nerve and associated macular findings. Review of the literature demonstrates that similar optic nerve findings observed have been variably described as optic nerve coloboma, morning glory disc anomaly, optic nerve excavation, and optic nerve pit, without a clear consensus. 3
The terminology used to describe this anomaly—whether coloboma, dysplasia, or morning glory disc anomaly—remains controversial. 10 We prefer the term papillorenal syndrome over the term “renal-coloboma syndrome,” given the lack of evidence that the optic nerve changes in this condition are attributed to a failure in the closure of the optic nerve disc fissure and therefore represent a true coloboma. A broader term such as “cavitary anomaly,” as previously suggested by other authors, may more appropriately encompass these findings. 11 Other cavitary optic nerve anomalies associated with maculopathy include optic nerve coloboma, morning glory disc anomaly, optic nerve pit, and staphyloma. 12
Furthermore, optic nerve manifestations in papillorenal syndrome can be quite variable, with some patients exhibiting a normal phenotype despite having a pathogenic PAX2 mutation. These findings may also be asymmetric and change over time. Our first case illustrates this asymmetry and the progressive evolution of cavitary optic nerve appearance over several years. Notably, the right eye initially demonstrated more subtle findings compared with the left eye, where surgical intervention may have contributed to the more pronounced cavitary changes observed from the initial presentation to the final follow-up (Figure 4).
The reported macular findings are also variable and may include macular pigmentation changes, macular or retinal detachment, retinoschisis, and maculopathy, among others. These differences may be attributed to the lack of OCT imaging in earlier reports.2–8
In our practice, we distinguish maculopathy from retinal detachment during slitlamp biomicroscopy by its confined central macular location and modest elevation. OCT demonstrates IRF extending toward the optic nerve, often associated with secondary SRF that remains predominantly restricted to the central macula, which we consider characteristic of this condition. In some cases, OCT may also demonstrate fluid tracking along the optic nerve, although this finding is not always observed.
In the second case, a diagnosis of morning glory disc anomaly was established. Although morning glory disc anomaly presents with a variety of clinical features, the associated retinal detachment typically differs from the maculopathy described in our cases. This detachment is typically more pronounced, extends beyond the macula, and lacks IRF. Similarly, retinal detachments associated with optic nerve coloboma originate from the intercalary membrane, are significantly elevated, and tend to extend inferiorly. Peripapillary choroidal neovascular membranes may also show similar macular findings; however, OCT and fluorescein angiography are typically diagnostic.
The patients responded well to our treatment protocol, which consisted of 3 key components:
The IRF and SRF do not typically resolve immediately after surgery, and close postoperative follow-up is required. In our experience with maculopathy associated with cavitary optic nerve anomalies, this fluid usually resolves within 6 months but may take up to 12 months or longer.
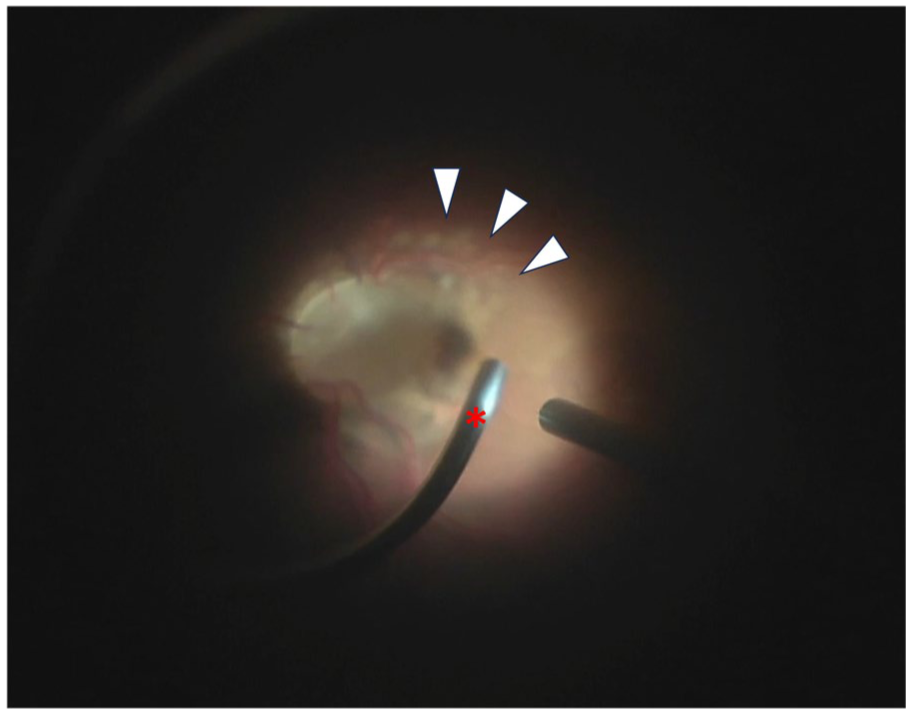
Intraoperative photograph of the right eye demonstrating peripapillary laser photocoagulation. White arrowheads indicate the the faint white laser spots applied to the temporal peripapillary retina. The red asterisk marks the laser probe directed toward the peripapillary region.
This structured surgical approach has proven effective in our patients, with no complications related to peripapillary laser treatment observed. Induction of a PVD may be the key surgical step, as vitreous traction on the anomalous optic nerve may facilitate the passage of fluid through the anomalous disc into the subretinal space. Previous reports describing subretinal migration of gas and silicone oil in patients with congenital cavitary optic disc anomalies provide clinical evidence of a structural defect within the anomalous nerve and suggest the presence of a communication between the vitreous cavity, subarachnoid space, and subretinal space. 13 This connection may be further addressed by applying peripapillary laser photocoagulation, which may help seal and maintain closure of the abnormal communication.
These cases highlight the importance of timely recognition and intervention to reduce the risk of vision loss. Although our cases did not exhibit the classic appearance of an optic nerve pit, they shared the characteristic retinal configuration associated with optic pit-like maculopathy. Sobol et al 14 reported a poor visual prognosis in untreated optic nerve pit maculopathy, with an average follow-up of 9 years, indicating that vision loss can occur within 6 months.
In our first case, delayed intervention in 1 eye contrasted with early laser treatment in the fellow eye, which maintained a BCVA of 20/20. The second case showed progression of maculopathy and worsening vision despite topical treatment, further emphasizing the potential need for surgical intervention in some cases. While untreated maculopathy may lead to vision loss, the ideal timing for intervention remains unclear. Additionally, genetic testing should be considered in patients with optic nerve cavitary anomalies and associated maculopathy, particularly when retinal vessels emanate from the optic disc rim rather than from the center of the disc, which often appears white or pale (Figures 4 and 6). These findings should raise suspicion for a PAX2 mutation and prompt assessment of personal and family history of renal disease. If a PAX2 mutation is confirmed, referral to nephrology is recommended.
The comparison between delayed and early interventions in our cases highlights the critical need for timely diagnosis and management to prevent vision loss. Despite these findings, the optimal timing and indications for intervention remain uncertain, and further research is necessary to standardize nomenclature and refine management strategies. Additionally, genetic testing should be considered in patients with cavitary optic nerve anomalies and associated maculopathy, as identification of a PAX2 mutation may provide further insights into the underlying etiology and guide individualized treatment strategies.
In conclusion, these cases underscore the complexity and variability in the diagnosis and management of optic disc pit-like maculopathy associated with cavitary optic nerve anomalies. The diversity in terminology and underlying mechanisms complicates treatment decisions. However, our structured surgical approach, involving careful induction of posterior vitreous detachment, peripapillary laser photocoagulation, and gas tamponade, was associated with favorable outcomes and no complications.
Footnotes
Ethical Considerations
This study was approved by the Institutional Review Board of the University of Illinois. All study procedures adhered to the tenets of the Declaration of Helsinki.
Consent to Participate
Written informed consent for participation was obtained from the patient’s guardians.
Consent for Publication
Written informed consent for publication of this case report and any accompanying images was obtained from the patient’s guardians.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.