
Abstract
X-ray crystallography is the most common technique for the determination of three-dimensional crystalline structures at the atomic scale. Since the discovery of the diffraction of X-rays by crystals over one hundred years ago, the technique has developed into an indispensable tool for material scientists and structural biologists worldwide. In this review, several milestones in the development of X-ray crystallography are presented, along with many of the Nobel laureates that made significant contributions to the success of the method. We conclude with a look at the current challenges in the field and speculate on the ensuing major developments that could lead to the next Nobel Prize related to X-ray crystallography.
Introduction
X-ray crystallography has been an essential technique for structural analysis over the last one hundred years. It has allowed us to view the world on a scale that was previously impossible, giving three-dimensional information and insight into the structural mechanisms and functions of important materials and biological molecules. These range from diamond and steel to the entire ribosome. The methods have become so widely used and automated that the various stages of the crystallographic pipeline have become increasingly optimized. High-throughput crystallization screening using robots allows scientists to test thousands of crystallization conditions with minimal effort. Multiple synchrotron facilities exist around the world, which generate bright sources of X-ray radiation suitable to obtain improved diffraction from smaller crystals. Automation of data collection and processing procedures has become the norm in the structure determination pipeline, abstracting the user from the complex intricacies of the diffraction data analysis. All of these aspects allow inexperienced users to solve fairly complex structures, making it a feasible and accessible tool for beginners. Structures that eluded determination a few decades ago have become routine today. Now crystallography is an indispensable tool and its use is crucial to several industries, including the mining, pharmaceutical, and even the aeronautical industry. The importance of X-ray crystallography is evidenced by the number of Nobel Prizes relating to the technique that have been won thus far.
However it is important to recognise that X-ray crystallography did not begin in its currently streamlined state. Incremental developments over the past hundred years have seen the technique advance beyond recognition. This review aims to highlight some of the important advances in X-ray crystallography over the last century, including the work for which several of the Nobel Prizes relating to X-ray diffraction were awarded.
The birth of a new scientific field: X-ray Crystallography
A new kind of ray
The story of X-ray crystallography begins with a professor at the University of Würzburg, Wilhelm Conrad Röntgen. As a child Röntgen did not show any notable outstanding academic ability, besides in mathematics (Simmons 2002), however he was very skilled in building mechanical contraptions, an attribute which proved particularly useful during his research career (Nobelprize.org 2014). Röntgen built a solid reputation as an outstanding experimental physicist with his independent investigations on the properties of gases and crystals. During the late 1800s he turned his attention to studying the properties of the rays (Glasser 1993) produced in vacuum vessels known as cathode ray tubes. However his experiments in 1895 led to unexpected results. Röntgen found that various materials such as paper, tin foil, wood, and several fluids were very transparent to a new kind of ray being produced from the tubes, which he termed X-rays ‘for the sake of brevity’ (Röntgen 1896). Furthermore, X-rays did not change direction in the presence of a magnetic field as did cathode rays (Dahl 1997). The significance of the properties of these rays was that they were almost immediately put to medical use to generate images of the interior of the human body (Simmons 2002) and Röntgen was awarded the first Nobel Prize in Physics in 1901 for his discovery. The X-rays were being produced by the acceleration of electrons and the same principle is used to generate X-rays at third generation synchrotrons today (Mitchell, Kuhn, and Garman 1999). However the origin of these rays was not the critical question in 1900, it was in fact their nature that caused significant debate amongst Röntgen's contemporaries.
X-rays: Wave or particle?
For centuries the question as to whether visible light was a particle or a wave had remained unanswered. Serious debate over the nature of light dates back to the seventeenth century when Isaac Newton had proposed a particle theory (or also commonly referred to as the corpuscular theory) of light. Opposition to the particle theory at the time largely came from Robert Hooke and Christaan Huygens who were refining the wave theory of light (Baierlein 1992). Over the next few centuries investigations by Thomas Young, and theoretical and experimental work by Augustin-Jean Fresnel along with James Clerk Maxwell's equations showing that electromagnetic waves travel at the speed of light, had all but confirmed their wave-like nature. However by the beginning of the twentieth century the wave theory of light was showing cracks. It failed to explain results from experiments carried out in 1887 by Heinrich Hertz and in 1902 by Philipp Lenard regarding the emission of electrons from metals using ultraviolet light, known as the photoelectric effect (Ewald 1962). Albert Einstein proposed a theoretical explanation of this phenomenon describing light quanta as a real physical entity, as opposed to just a theoretical concept as perceived by Max Planck (Pais 1979), for which Einstein received the Nobel Prize in Physics in 1921.
At this point however, it was not clear whether X-rays were even related to visible light (Stuewer 1971) let alone whether they were waves or particles (Holton et al. 2014). In 1906 Charles Glover Barkla had shown that X-rays could be polarized and this became key evidence that they were wave-like in nature. However due to Sir William Henry Bragg's work on radioactivity at the University of Adelaide, the very same evidence that was seen to support the wave theory of light could also be interpreted by a corpuscular description of X-rays (Stuewer 1971). It would take intuition from Max von Laue to add indisputable evidence for the wave-like nature of X-rays.
Max von Laue's academic pursuits generally swayed towards fundamental physical issues so for this reason a PhD student, Paul Peter Ewald, visited Laue to seek assistance on his thesis topic. It was based on how light waves interact with a lattice of polarizable atoms and Ewald had drawn unexpected conclusions from his results. Although Laue could not help Ewald he was nonetheless intrigued by a problem arising from Ewald's work and wanted to know the effect of passing light with a lower wavelength through the lattice (Ewald 1962). Very soon afterwards Laue performed experiments in collaboration with Walter Friedrich and Paul Knipping in which they passed X-rays through crystals of copper sulphate, producing diffraction patterns (Friedrich, Knipping, and Laue 1913) proving the wave-like nature of X-rays and validating the space lattice description of crystals (Eckert 2012). Max von Laue was awarded the Nobel Prize in 1914 for discovering the diffraction of X-rays by crystals (Nobelprize.org 2015b). The distinguished polymath, Henri Poincaré, is reported to have said ‘if the value of a discovery is to be measured by the fruitfulness of its consequences, the work of Laue and his collaborators should be considered as perhaps the most important of modern physics’ (Stuewer 1971). Unfortunately Poincaré died in the summer of 1912 and so did not live to see the veracity of his statement. It was only one year after Laue's discovery that a father and son duo based in England made the next giant leap, and their discovery would change the understanding of the world as we knew it.
Creation of a new scientific discipline
Although Friedrich, Knipping, and Laue had successfully demonstrated X-ray diffraction, their initial interpretation of the phenomenon was incorrect. As noted by a young Henry Moseley in a letter to his mother in November 1912, ‘the men who did the work entirely failed to understand what it meant, and gave an explanation which was obviously wrong’ (Heilbron 1974). University of Leeds Cavendish Professor of Physics, William Henry Bragg (WHB), and his son William Lawrence Bragg (WLB) were well aware of Laue's misinterpretation and Moseley writes that he already knew that ‘Bragg and his son had worked out an explanation a few days before us … We are therefore leaving the subject to them’.
Being a supporter of his father's views, WLB tried, unsuccessfully, to explain Laue's results using a corpuscular theory of X-rays. Increasingly he became convinced that an explanation was only possible via a wave description. Laue had proposed that the diffraction pattern was a result of the diffraction of certain wavelengths of the X-ray beam by the crystal lattice (Ewald 1962). However WLB noticed that the reflections became more elliptical in shape the further away the plate was placed from the sample. WLB's model of this phenomena can be seen in Figure 2 of a paper entitled ‘The diffraction of short electromagnetic waves by a crystal’ (Bragg 1913a). This phenomenon could not be explained by Laue's ideas but instead by the idea that X-rays were being reflected at various angles by planes of atoms in the crystal. It was WLB who proposed the famous equation that relates the distance, d, between planes of atoms in a crystal to the position of a reflection on a detector via the angle of reflection, θ, which is known as Bragg's Law
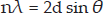
where λ is the wavelength of the incident radiation and n is an integer denoting the number of wavelengths difference in path length for X-rays that have been reflected from successive planes. Using this new model of X-ray diffraction, WLB managed to solve the structure of the zinc blende crystal (Bragg 1913a). William Pope, Professor of Chemistry at Cambridge, encouraged WLB to solve the structures of other crystals including NaCl and KCl (Figure 1) (Bragg 1961).
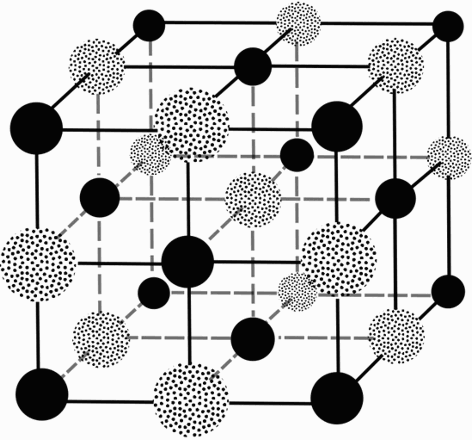
Face centred cubic lattice of NaCl. Solid spheres represent sodium atoms, dotted spheres represent chlorine atoms.
It should be noted that although WLB used a wave description of X-rays to resolve Laue's experimental results, both Braggs still maintained the view that the particle description of X-rays was still necessary to explain some experimental observations. In response to Laue's results WHB remarked ‘the problem then becomes, it seems to me, not to decide between two theories of X-rays, but to find… one theory which possesses the capabilities of both’ (Ewald 1962). The issue would not be resolved until Arthur Compton's discovery of the ‘Compton effect’ in 1922, which demonstrated the particle nature of elctromagnetic radiation (Stuewer 1971).
Despite WLB's efforts to solve crystal structures, it was not until WHB built the first X-ray spectrometer and coupled this with an ion chamber to measure the strength of individual reflections, did crystal structure analysis really take off. The Braggs worked together throughout the summer of 1913 and made many discoveries. WHB was more interested in X-rays than crystal structures and he discovered that metals give X-rays with characteristic energies (Ewald 1962), a property that is exploited today in microbeam proton induced X-ray emission (microPIXE) to quantitatively determine the metal content in proteins (Garman and Grime 2005). WLB was generally interested in solving crystal structures and a series of papers from the Braggs were published over the next two years (Bragg 1914b, Bragg and Bragg 1913, Bragg 1913b, Bragg 1914a). The Braggs had opened up the field of structural analysis and were awarded the 1915 Nobel Prize in Physics for services in the analysis of crystal structure by means of X-rays (Nobelprize.org 2015c).
Early Developments and Pioneers
The years 1912–1920
The years from 1912 to 1920 saw many advances in experimental X-ray diffraction methods and theory. No attempt at an exhaustive list will be made here, but instead some highlights are provided. The concepts of the reciprocal lattice and the sphere of reflection (more commonly referred to as the Ewald sphere) were first conceived and published in 1913 (Ewald 1913). Importantly they allow for a visual construction, albeit theoretical, through which it is possible to determine when the diffraction conditions will be satisfied for a given crystal lattice. This allows accurate prediction of the positions of reflections on a detector. Charles G. Darwin's papers on calculating the total intensity of a reflection, published in two parts in 1914 (Darwin 1914a, Darwin 1914b), is one of three papers regarded by WLB as ‘standing out from the rest’ during this period (Bragg 1961). It was a treatment that covered both perfect and imperfect crystals which helped to understand how the intensity of a recorded reflection depends on the parameters of the apparatus, the incident beam intensity, the physical constants and the structure of the crystal (Holton et al. 2014). The other two papers that were especially noted by WLB were by his father, in which WHB first suggested and experimented with recording reflections in an exposure during which the crystal is oscillated (Bragg 1914c). The intensity then can be measured by integrating it over the reflection. This method of data collection has been standard practice for crystallography experiments since then. The second paper is from the 1915 Bakerian Lecture given by WHB (Bragg 1915) in which he suggested that structure solution could be possible through Fourier analysis, despite the method being in its infancy at the time. Nowadays structure solution methods would be unthinkable without Fourier analysis, due to the complex nature of the biological molecules that are under study.
Peter Debye
Alongside the above advances, another pioneer was making significant headway in extending knowledge on molecular structure. Peter Josephus Wilhelmus Debye, primarily a physicist, made several contributions to the field of physical chemistry. In particular his work on dipole moments provided a means to obtain accurate estimates of the structure of gases and liquids (Davies 1984). More specifically relevant to X-ray diffraction, Debye investigated the effect of atomic vibrations in the crystal lattice on the diffraction pattern (Eckert 2011). Prior to Laue's experiment Debye is said to have told Laue that atomic vibrations of the atoms within the crystal would obscure any interference. Only a few months after Laue's successful experiments, Debye showed that the intensity of a reflection is reduced by a factor of
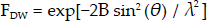
where B is a factor proportional to the mean-square displacement of the scattering atom and the other terms are as in equation (1) (Frauenfelder 1989). This result, known as the Debye-Waller factor, was experimentally validated by WHB in 1914. In 1916 Debye and Paul Scherrer developed the technique of powder crystallography (Davies 1984). The advantage of this method is that it does not require relatively large single crystals as did the X-ray spectrometer which was in widespread use at the time. Powder crystallography is still in use today and current developments are allowing further structural insight on macromolecules (Margiolaki and Wright 2007). In 1936 Debye was awarded the Nobel Prize in Chemistry ‘for his contributions to our knowledge of molecular structure through his investigations on dipole moments and on the diffraction of X-rays and electrons in gases’ (Nobelprize.org 2015a).
The years 1920–1930
Improvements in the accuracy of intensity measurements coupled with validation of Darwin's work led to the analysis of more complex inorganic crystals during the 1920s. Prior to this period, organic crystals were considered too complex, and structure solution required prior structural knowledge from studies within organic chemistry. Towards the end of the decade, direct structure solution of organic crystals became feasible (Bragg 1961). Notably, in 1929 Kathleen Lonsdale was able to show that the benzene ring in hexamethylbenzene is hexagonal and planar, and that the ring is similar in both its structure and size to the graphite ring (Lonsdale 1929). Finally Fourier analysis techniques were beginning to take form, setting the stage for the elucidation of ever more complex crystal structures.
James Sumner
Thus far most major developments within X-ray crystallography had been concerned with the improvements of the experimental apparatus or the analysis of the data. However an essential aspect of the technique, and arguably the most difficult, is the ability to obtain crystals. Crystals of small inorganic and organic molecules are, in general, relatively easy to acquire compared with crystals of macromolecules such as proteins and carbohydrates. In 1926 it was the work of a relatively unknown biochemist that would dramatically improve crystallization techniques for proteins.
James Sumner led a busy life juggling teaching with research at Cornell University. The research facilities were primitive, he only had one graduate assistant and a lack of research funding, but this did not deter him from aspiring to make a great scientific discovery. In 1917 Sumner decided that he wanted to isolate an enzyme, a feat that had not been achieved before. He chose urease as his target, because he hoped it would be a globulin; globulins are known to precipitate easily by dialysis, and also since it was expected to be one enzyme instead of a mixture, it would be easy to quantify (Sumner 1926). After nine years Sumner eventually managed to obtain crystals of urease by dissolving jack bean meal in 32 per cent acetone instead of alcohol, filtering the solution and leaving it overnight in an ice chest. Although the crystallisation was significant in its own right, the real controversy came from interpretation of the subsequent analysis. In the morning Sumner saw octahedral crystals and decided to redissolve them in a solution which subsequently gave typical results characteristic of a protein (Edsall 1976). At the time Sumner's results were met with skepticism, since the majority of the biochemical community, most notably Nobel Laureate Richard Willstätter, regarded enzymes as chemical substances separate from proteins. It was only when John Northrop proved Sumner's hypothesis about the nature of enzymes in 1935 that it was generally accepted that enzymes were proteins (Brock 2001). Sumner and Northrop shared the 1946 Nobel Prize in Chemistry along with Wendell Stanley for their work on enzymes.
The 1930s onwards
Structure solution of ever more ambitious molecules was being attempted from the 1930s onwards. This meant that a growing number of parameters for an increasing number of atoms were having to be determined. The trial and error method of placing atoms in the unit cell and comparing the results from the model structure with the data was becoming infeasible. More information was necessary to tackle the growing challenge of structure determination. Fourier analysis was a promising method for structure determination because it eliminated the aspect of trial and error. The problem was that knowledge of the phases of the X-rays diffracting from the crystal was required but could not be measured directly in the experiment. This is known as the phase problem. Despite this, new ways of manipulating the Fourier series became useful for X-ray analysis. In 1934 Arthur Lindo Patterson published his ground breaking paper where he introduced the Patterson function (Patterson 1934), which gives the interatomic distances within the molecule without any knowledge of the actual phase contributions from each term in the Fourier series. This meant restraints could be applied to the positions of atoms in the unit cell using the data collected directly from the X-ray crystallography experiment. In 1937 Monteath Robertson and Ida Woodward published a paper on the structure of nickel phthalocyanine (Robertson and Woodward 1937). This achievement was made possible by a mixture of Fourier analysis and direct determination of the phases by comparison of the reflection intensities with those of free phthalocyanine (without bound nickel), in a similar manner to the isomorphous replacement method (section ‘The first protein structure’ (below)).
Further to the advances in X-ray analysis of organic and inorganic chemical molecules, Dorothy Hodgkin (née Crowfoot) highlights 1934 as the second of three dates of special importance for protein crystallography (the first date being the first X-ray diffraction pattern by a crystal in 1912 by von Laue) (Hodgkin 1979). John Desmond Bernal and Hodgkin took the first X-ray diffraction images of a protein in its mother liquor inside a capillary tube (Bernal and Crowfoot 1934). Previously protein crystals were exposed to air prior to X-ray exposure, which dehydrated the protein crystals and resulted in no observable diffraction. This breakthrough meant that a method of performing X-ray analysis on crystals of proteins was now available. This was the beginning of macromolecular X-ray crystallography.
The advent of biological crystallography
The structure of DNA
By the early 1950s the study of deoxyribonucleic acid (DNA) already had a long history. In the 1860s Gregor Mendel had studied genetic inheritance investigating the phenotypes of several generations of pea plants (Bateson and Mendel 2013). Later in the same decade Friedrich Miescher had managed to isolate DNA, which he called nuclein at the time, from a variety of different cells (Dahm 2008). Experiments deducing the importance of genes in biochemical pathways and the effect of genetic mutations were carried out in subsequent decades. These discoveries were evidence for the fact that genes were responsible for genetic inheritance and the information was encoded inside almost every cell in higher organisms. During the 1930s DNA could not be crystallized so X-ray diffraction images were generated by irradiating fibres of DNA. William Astbury along with his PhD student, Florence Bell, were the first to take X-ray diffraction images from DNA (although they believed DNA was a nucleoprotein complex). They calculated that successive nucleotides in a nucleic acid chain were about 3.3 Angstroms apart (Astbury and Bell 1938). However these diffraction images were not of great quality so it would not be until Rosalind Franklin and Maurice Wilkins began their research into DNA that its structure would be revealed.
Maurice Wilkins moved to King's College London in 1946, working under John Randall, who became Head of Physics there. One of their first achievements was to build reflecting microscopes for ultraviolet light that were used to show that genes were made of DNA (Wilkins 2003), not protein as many had postulated. In 1950, Wilkins worked with a research student, Raymond Gosling, and managed to obtain good diffraction patterns from DNA fibres. However they needed sharper patterns to obtain sufficiently detailed information on the structure. This would require specially designed X-ray equipment and someone skilled enough to operate it (Wilkins 1995).
Rosalind Franklin spent a few years in Paris working in a French government crystallographic laboratory on coal before moving to King's College London in 1951 (Maddox 2003). Franklin was brought in to get the new X-ray equipment up and running but was also assigned to work on DNA, since Wilkins was away during her arrival. The relationship between Wilkins and Franklin was strained from the start and after a ‘big blowup… they effectively didn't communicate’ (Watson 2008). Despite the hostile environment (Gibbons 2012), Franklin managed to generate significant data with Gosling. Together they collected X-ray diffraction data from both A and B forms of DNA that would be vital to the solution of its structure. Before Franklin was ready to publish her results, Wilkins presented Franklin's data to James Watson, a young researcher at Cambridge, including the famous photo 51 (Figure 2), without Franklin's knowledge. These data, along with prior information from DNA studies allowed Watson and Francis Crick to solve the structure of DNA, which they subsequently published (Watson and Crick 1953). In the years following, Wilkins and others spent much time improving the diffraction patterns to validate the proposed model of DNA. Watson, Crick and Wilkins were jointly awarded the Nobel Prize in Physiology or Medicine in 1962 ‘for their discoveries concerning the molecular structure of nucleic acids and its significance for the information transfer in living material’ (Nobelprize.org 2015e). Franklin tragically passed away in 1958 aged 37 from ovarian cancer and so was not awarded the Nobel Prize (it is not awarded posthumously). However her notebook has shown that as well as obtaining significant experimental results via X-ray diffraction, she had also correctly deduced many properties of the DNA molecule (Klug 1974) suggesting she too was incredibly close to independently discovering its structure.
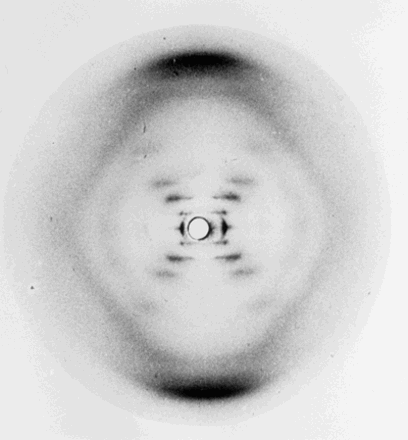
Photo 51. Fibre diffraction image of the B form of DNA taken by Rosalind Franklin and her PhD student, Raymond Gosling in 1952.
The first protein structure
Analysis of diffraction patterns from protein crystals was still a complex and unsolved problem. The number of reflections in a diffraction pattern from a protein crystal typically range in the thousands. The ionization spectrometer was not suited to efficiently record this number of reflections. The Weissenberg camera, which was developed in 1924 (Weissenberg 1924), became much more widely used for recording reflections from diffraction patterns, particularly after Bernal had described how to interpret the rotation photographs it generated (Bernal 1926). However the big question for protein crystallography was how can the phase of each reflection be directly determined from the X-ray diffraction data? Given that a protein's function is predominantly determined by its structure, the answer to this question would be a monumental step towards the goal of understanding how living organisms function on a molecular scale.
In 1937 Max Perutz began work on X-ray analysis of crystalline proteins under Bernal at the Cavendish Laboratory in Cambridge. Perutz managed to obtain diffraction images of haemoglobin, a type of globular protein that is found in red blood cells responsible for oxygen transport. Initially Perutz investigated solving the phase problem by analysing the changes to the intensities (and positions) of reflections in the diffraction pattern for different shrinkage states of the crystals while they were drying (Hodgkin 1979). This turned out to be extremely difficult and required highly accurate intensity measurements. However WLB was very supportive of Perutz's work and persuaded the Medical Research Council to establish a new laboratory for the study of biological systems at Cambridge to support it (Dickerson 2008). John Kendrew began research with Max Perutz as a PhD student in 1945 and worked on myoglobin, a protein which is roughly a quarter of the size of haemoglobin. Eight years later Perutz would make a discovery that would become the third significant milestone in the development of protein crystallography (Hodgkin 1979). In 1953 Perutz managed to successfully use the method of isomorphous replacement to solve the phase problem for the haemoglobin diffraction patterns. Mercury benzoyl atoms were incorporated into a haemoglobin crystal without changing the atomic arrangement of the protein. Upon irradiation with X-rays, the mercury benzoyl derivative crystal gave a diffraction pattern with measurable differences in the intensities of reflections, which were used to locate the positions of the heavy atoms. In 1957 Kendrew solved the first protein structure, myoglobin, by X-ray diffraction analysis, and by 1959 he had achieved atomic resolution of the myoglobin structure. In the same year Perutz solved the structure of haemoglobin (Figure 3) (Dickerson 2008, Thomas 2002). In 1962 Kendrew and Perutz were awarded the Nobel Prize in Chemistry for their studies of the structures of globular proteins.

Max Perutz with a balsa wood model of the structure of haemoglobin, the protein in red blood cells. This model was built by Max Perutz in 1959.
The immediate consequences of the first protein structure determination
The structures of several protein molecules were solved following the achievements of Kendrew and Perutz (North and Phillips 1969). Notably the structure of the first enzyme, lysozyme, was determined in 1965 (Blake et al. 1965) which allowed scientists to propose a mechanism for its function at the atomic level. However, the X-ray analysis of protein crystals presented huge obstacles. The number of parameters required to solve protein structures soared (Bragg 1961) and the calculations required for the structure solution were intractable by humans so the use of computers became necessary for processing the data. Additionally, the use of one heavy atom derivative to determine the phases of the reflections in the diffraction pattern (single isomorphous replacement) led to an ambiguity in the possible phases of the reflections. Perutz overcame this problem by creating multiple heavy atom derivatives (multiple isomorphous replacement). Johannes Bijvoet suggested an alternative method to solve the phase ambiguity that avoided the problem of having to find multiple suitable derivative atoms. Bijvoet suggested that choosing an X-ray wavelength similar to the absorption edge of the heavy atoms would change the intensity of certain reflections (Bijvoet 1954). This phenomenon, known as anomalous scattering, effectively acts in a similar way to having another heavy atom derivative. The additional intensity changes could be used to solve the phase ambiguity. This method was first used by Gopalasamudram Ramachandran in 1956 for the structure of L-ephedrine hydrochloride, and for protein structure determination by David Blow in 1958 (Blow and Rossmann 1961). The groundwork for another phasing method was first published by Blow and Michael Rossman in 1962 when they proposed a method for finding non-crystallographic symmetry from the reflection intensities (Rossmann and Blow 1962). With the creation of the Protein Data Bank (PDB) (see section Technological developments in the 1970s) and solution of many more protein structures, the method was developed into what is now known as molecular replacement. This method allows the phases of an unknown protein structure to be estimated given a known, partially homologous structure. Currently almost 70 per cent of macromolecular structures are solved using the molecular replacement technique (Evans and McCoy 2008).
Dorothy Crowfoot Hodgkin
Dorothy Crowfoot Hodgkin was one of the most significant pioneers of X-ray crystallography. In particular her desire and perseverance to tackle complex molecules led to her producing some of the most ambitious and ground breaking structures of her time.
Hodgkin began work in crystallography in 1932 under the supervision of Bernal. She was involved in obtaining the X-ray photography of pepsin in 1934, but also succeeded in solving the structure of cholesterol (Hodgkin 1965). Later Hodgkin was approached to determine the structure of penicillin. Its antibacterial properties were already documented but during the Second World War it was showing promise in fighting bacterial infections contracted by humans. Determination of its structure would provide a means for production of synthetic penicillin and became a top priority during the war. Hodgkin achieved this in 1945, although due to wartime secrecy, the results were not published immediately (Crowfoot et al. 1949). At the time, penicillin (23 non-hydrogen atoms) was the largest molecule solved by X-ray crystallography.
Hodgkin also started working on the structure of vitamin B12 after crystals of the molecule were given to her in 1948, and in 1956 she determined it (Hodgkin et al. 1956). Hodgkin received the Nobel Prize for Chemistry in 1964 ‘for her determinations by X-ray techniques of the structures of important biochemical substances’ making her the only female British recipient of this award. However this was not the last of Hodgkin's major contributions to determining the structures of medically important molecules. In 1969, more than thirty years since the initial X-ray photograph was taken, Hodgkin solved the structure of insulin (Kass-Simon, Farnes, and Nash 1993).
Technological developments in the 1970s
It was anticipated that structure determination of biological molecules would increase over the coming years, so in 1971 Walter Hamilton led the creation of the PDB at Brookhaven National Laboratory. This was a free, open access repository to store information on the structures of the macromolecules that were solved. It was the first of its kind and a huge achievement for the crystallographic community. Beginning with only 7 structures in 1971 the PDB now contains 105,906 structures (20 January 2015) with the number increasing rapidly every year (Berman et al. 2000). Synchrotron radiation sources, where radiation is emitted from highly energetic electrons being deflected in a roughly circular path by a magnetic field, began to be used for protein crystallography experiments in 1974 (Mitchell, Kuhn, and Garman 1999). Synchrotrons are crucial to the study of protein structures at atomic resolution because the greater X-ray beam flux increases the intensity of reflections in the diffraction pattern. Additionally, the ability to tune the wavelength of these X-rays has allowed crystallographers to take advantage of anomalous dispersion to solve the phase problem (Lindley 1995). Photographic film was replaced by electronic area detectors since they provide better dynamic range, improved information per photon and better time resolution (Schulz and Rosenbaum 1978). Georges Charpak was awarded the Nobel Prize in Physics in 1992 ‘for his invention and development of particle detectors, in particular the multiwire proportional chamber’ and this technology was subsequently used in detectors for macromolecular crystallography. Developments in the experimental hardware meant that improvements to the existing crystallographic software became a necessity to process the vast amounts of data being generated. The Collaborative Computational Project Number 4 (CCP4) in protein crystallography was set up in 1979 to establish a collection of software that could be used by crystallographers for processing diffraction data. This project still exists and distributes its software to academic and commercial users worldwide (Winn et al. 2011).
The development of electron crystallography
Alongside the developments of X-ray diffraction techniques, the electron diffraction phenomenon was also being exploited for structural analysis investigations, since the discovery that electrons could be diffracted by crystals (Nobelprize.org 2015d). Electron microscopy can be performed on samples unsuitable for X-ray crystallography (Crowther and Klug 1975). Since electrons interact more strongly with atoms than X-rays, the crystal samples for electron microscopy are much thinner than those used in X-ray crystallography. However this can lead to problems such as increased radiation damage and loss of three-dimensional order (Unwin and Henderson 1975). Aaron Klug made a major breakthrough by tackling the image reconstruction problem inherent in the images obtained by electron microscopy. By applying two-dimensional spatial filtering and three-dimensional image reconstruction, Klug was able to interpret the data and combine them with X-ray diffraction data to determine the three-dimensional structure of biologically important molecules such as viruses, chromatin and the nucleosome (Klug 1983). For this work Klug was awarded the Nobel Prize in Chemistry in 1982.
Direct methods for solving the phase problem
In 1985 Herbert A. Hauptman and Jerome Karle shared the Nobel Prize in Chemistry ‘for their outstanding achievements in the development of direct methods for the determination of crystal structures’. The term direct methods is used to refer to the fact that the crystal structure is determined directly from the diffraction data. Although the foundations for this work were laid in the early 1950s, scepticism about the validity of direct methods was a major reason why the results were not utilized immediately (Hauptman 1991). It was only after techniques for direct methods were incorporated into computer programs, and complex structures throughout the 1960s and 1970s were solved using these methods, that the crystallographic community recognised their power. Using only the assumption that the electron density is positive and atoms are discrete in a crystal, the phase problem becomes over determined in solvable cases and hence the diffraction data become sufficient for structure determination. The derived triplet relation states that the phase of a particular reflection can be determined from the knowledge of the phases of two different reflections (Hauptman and Karle 1953). Direct methods are useful for small molecule crystals but generally for protein crystals, the resolution required is unattainable for direct methods to be appropriate (Taylor 2010).
Membrane protein crystallography
All of the protein structures solved before 1975 were of soluble proteins. However, membrane proteins represent around a quarter of the proteomes of most organisms (Carpenter et al. 2008), but they are difficult to crystallize because they are particularly insoluble. This means expression, purification, and crystallization techniques generally have to be modified to generate suitable crystals for X-ray analysis. The first low resolution membrane protein structure was solved by Richard Henderson and Nigel Unwin in 1975 by electron diffraction, and the first at high resolution, the photosynthetic reaction centre, was solved by Johann Deisenhofer, Hartmut Michel, and Robert Huber in 1985 using X-ray diffraction (Deisenhofer et al. 1985). This was a significant achievement for several reasons: it demonstrated that experimental methods could be manipulated to produce suitable crystals of membrane proteins for X-ray crystallography experiments, it was the largest asymmetric structure to have been determined by X-ray crystallography (Vinothkumar and Henderson 2010), and finally it represented a huge step in the understanding of one of the most important chemical processes on Earth, photosynthesis. For their work Deisenhofer, Michel, and Huber were awarded the Nobel Prize in Chemistry in 1988.
Third generation synchrotron sources and radiation damage
In the 1990s, several third generation synchrotron sources became operational, increasing the brightness of the available incident X-ray beams which caused irradiated crystals to routinely suffer radiation damage. This phenomenon is recognized as one of the major obstacles in protein structure determination (Garman 2010). Cryo-protection techniques to reduce the effects of radiation damage were introduced in the late 1980s and early 1990s (Garman and Schneider 1997, Hope 1988, Teng 1990) and the majority of X-ray crystallography diffraction experiments now take place with the crystals held at around 100K. The advent of third generation synchrotron sources along with cryo-crystallography saw an exponential increase in the number of structures deposited to the PDB: 507 in 1990 to over 10,000 in 2000 (Berman et al. 2000).
Important biochemical structures
A combination of advances in molecular biology techniques for protein production, robots to aid in the screening of many possible crystallization conditions, tuneable intense synchrotron X-ray sources, detector technology, crystallographic software with new algorithms based on maximum likelihood methods, computing power and experimental methods, has increasingly made structure solution of relatively complex molecules more routine than ever before. This has paved the way for scientists to begin solving molecular structures that are not only biochemically important but were also previously viewed as impossible. In 1997 Paul Boyer and John Walker shared the Nobel Prize in Chemistry ‘for their elucidation of the enzymatic mechanism underlying the synthesis of adenosine triphosphate (ATP)’, the energy currency of biology. This was followed in 2003 by Rob MacKinnon's Nobel Prize in Chemistry award for his work on the structural and mechanistic studies of ion channels. Soon after, Roger Kornberg was awarded the Nobel Prize in Chemistry ‘for his studies of the molecular basis of eukaryotic transcription’ in 2006. The structure determination of the ribosome was a huge achievement for crystallographic methods, since it consists of around 280,000 non hydrogen atoms compared with the 1260 in myoglobin solved in 1957 (Figure 4) (Garman 2014). For their ‘studies of the structure and function of the ribosome’ Venekatraman Ramakrishnan, Thomas Steitz, and Ada Yonath shared the 2009 Nobel Prize in Chemistry. In 2012 Robert Lefkowitz and Brian Kobilka shared the Nobel Prize in Chemistry ‘for studies of G-protein-coupled receptors’, an integral membrane protein family that account for about 40 per cent of current pharmaceutical drug targets (Filmore 2004).
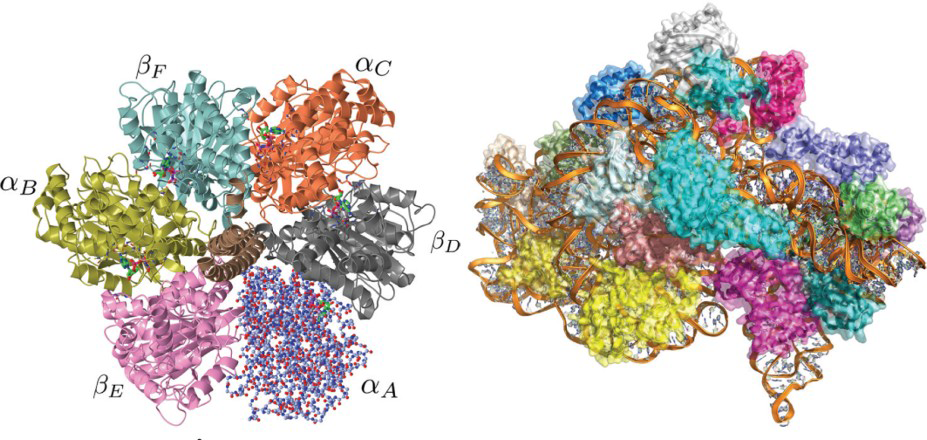
(left) 2.8Å structure of F1-ATPase from bovine heart mitochondria (PDB code 1BMF). The 393.9kDa structure consists of 6 alternating α and β subunits arranged in a circle approximately 100Å across surrounding a central γ subunit (brown). It is part of the ATP synthase enzyme that builds adenosine triphosphate (ATP), the molecule that powers the majority of cellular processes. The subunit labelled αA is shown as a more realistic atomistic representation. This type of molecular representation is generally harder to interpret and hence the ribbon representation depicted by the rest of the structure is more commonly used in displaying molecular structures. (right) 3Å structure of the 30S subunit of the ribosome from Thermus Thermophilus bacterium (PDB code 1J5E). The molecular weight of this structure is approximately 786.2kDa and consists of 20 separate proteins surrounding a ribonucleic acid (RNA) molecule shown in orange. The ribosome is a large nucleoprotein complex that translates the information encoded within the RNA into proteins.
Conclusions
Over the past hundred years, X-ray crystallography has transformed our understanding of the materials around us and of the molecules within us. For example, the field has elucidated the properties of chocolate and the structure of turbo-engine blades, as well as enabling the discovery of the anti-influenza neuraminidase inhibitors, Relenza and Tamiflu. Crystallography (powder patterns) was even used in 2012 by NASA's Curiosity rover to analyse samples of soil on Mars (Bish et al. 2013). It has become a routine tool amongst the growing arsenal of structural and biophysical characterisation methods, and can now be accessed even by a beginner. It would be naïve to think that crystallography has currently reached the limit of its capabilities: the real question is what will the next major developments be? Several different areas of the field are currently attracting excited attention.
Firstly, membrane proteins constitute a high percentage of pharmaceutical drug targets but working with them is still challenging at many stages of the structure determination pipeline. However, the number of unique membrane protein X-ray structures deposited in the PDB is currently rising exponentially and is now over 500, having only been approximately 50 in the year 2000. A new method of crystallizing membrane proteins in lipidic cubic phase (LCP) is contributing to this success, as well as the availability at synchrotrons of the intense small (several micron) X-ray beams necessary in order to locate crystals in the opaque LCP medium. Further development in the experimental methods for investigating membrane proteins can be expected in the future.
Secondly, electron microscopy (EM) has traditionally been used to study the overall shapes of large molecules and complexes, since the attainable resolution of the data, as well as the minimum tractable size of complexes, have both been limiting factors. However, due to very recent advances in technology which has seen the introduction of direct complementary metal-oxide-semiconductor detectors, cryo-EM techniques both for diffraction experiments and for single particle imaging are now approaching the resolution of X-ray data (e.g. the 70S ribosome at 4 Å (Grigorieff 2013)), and the latter method obviates the limiting requirement for a crystal. Thus we are likely to see a large increase in the number of interesting structures being solved by EM.
Thirdly, radiation damage is still a major limiting factor in crystallographic macromolecular structure solution. The main method employed to reduce the effects of radiation damage is currently cryo-crystallography, although dose spreading strategies can also be employed to good effect (Zeldin, Gerstel, and Garman 2013). However, recent developments in serial femtosecond crystallography (SFX) using X-ray free electron lasers (XFELS) have shown promising results in reducing or even totally avoiding radiation damage and also in generating interpretable diffraction from membrane proteins. Significant improvements in the experimental setup and data analysis are required before this method can become a routine and generally accessible technique for structural analysis (Chapman et al. 2011). In the future it may even be possible to carry out single particle imaging with XFELs.
In addition, vast increases in computational capability now enable much more powerful algorithms to be implemented both in the bioinformatics arena and in efforts to predict protein structures solely from their sequences. The latter may eventually remove the need for crystals or real experimental X-ray data.
Recent structural genomics efforts have resulted in new technology which is benefitting the entire field of crystallography but the function of many of the solved molecules is unknown. A challenge for the future is not only to understand their functions, but also to undercover their dynamical movement and thus their mechanisms.
These are few areas in which developments in structural analysis are still pushing the boundaries of the possible. It is not a matter of ‘if’, but rather ‘when’, the next major breakthrough in crystallography will lead to another Nobel Prize for work relating to this diverse and exciting field of study.
Footnotes
Acknowledgements
We sincerely thank Charles Bury for his help in making the figures and we gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC) in the UK for funding through a studentship at the Systems Biology programme of the University of Oxford Doctoral Training Centre (JBB).
Notes on contributors
Jonathan Brooks-Bartlett is a DPhil student at the University of Oxford, UK. He studied Mathematics at the University of Southampton, specializing in applying mathematics to biological problems, before moving to Oxford to work with Elspeth in the Department of Biochemistry. His research focusses on the study of radiation damage in protein crystallography experiments with a view to correcting the damaged data. Jonathan is also a keen science communicator and works with the British Crystallographic Association's Education and Outreach team to deliver public engagement activities throughout the UK.
Correspondence to: Jonathan Brooks-Bartlett, Department of Biochemistry, University of Oxford, South Parks Road, Oxford, OX1 3QU, UK. E-mail:
Elspeth Garman is Professor of Molecular Biophysics at the University of Oxford, UK. She started her working life aged eighteen as a volunteer teacher in Swaziland, Southern Africa. Following a degree in Physics at Durham University, she did a DPhil in Experimental Nuclear Structure Physics at Oxford. After seven years as a Nuclear Physics Research Officer and Tutor, she changed fields to protein crystallography. Her main research interests are in improving methods for finding the three-dimensional shapes of medically important biological molecules so that larger and more complicated structures can be determined, and disease pathways can be understood at a molecular level.
Correspondence to: Elspeth Garman, Department of Biochemistry, University of Oxford, South Parks Road, Oxford, OX1 3QU, UK. E-mail: