
Abstract
Localised corrosion is a cause of unanticipated and sometimes catastrophic failures of equipment, transport vessels and infrastructures. Therefore, the development of modern corrosion-resistant materials and inhibitors by design is both technically and economically attractive. In the coming decades, industrial components will be engineered from molecular structures. This prospect provides the designer with a truly enormous range of choices in design, which is a situation that demands predictive tools that can link molecular structures with the final component performance. In particular, the development of alloys and inhibitors can replace the use of toxic compounds in protecting metal surfaces. To execute a tailored design programme, it is necessary to understand how corrosion and the associated processes occur from the molecular level to the component level and how the overall system behaviour emerges because of the inherent links among different scales. Therefore, in the present work, the literature on theoretical modelling of localised corrosion and related experimentation are reviewed from a multiscale viewpoint. The review addresses (a) the challenges in the theoretical formulation of the important phenomena that influence localised corrosion and (b) the hurdles facing computational methods. It is shown that (i) the existing models lack the resolution to design effective corrosion-resistant systems, (ii) the numerical strategies for linking the scales are in a state of evolution and (iii) there are gaps in the experimental characterisation of the corrosion system, particularly at the lower end of the scales. Suggestions are provided towards the construction of a multiscale model (MSM) for localised corrosion.
List of abbreviations
artificial neural networks
boundary element method
balance volume
cellular automaton
generalised gradient approximation
critical crevice solution theory
critical solution chemistry theory
control-volume finite element method
density functional theory
dissipative particle dynamics with energy conservation
finite difference method
finite element method
finite volume method
high field model
IR drop theory
kinetic Monte Carlo
inear combination of atomic orbitals
Local Spin Density Approximation
lattice material point method
Monte Carlo
molecular dynamics
multiscale model
open circuit potential
probability density function
quantum mechanics
quantitative structure activity relationship
Spanish initiative for electronic simulations with thousands of atoms
smoothed particle hydrodynamics
Vienna ab initio simulation package
X-ray photoelectron spectroscopy
Introduction
Localised corrosion is the accelerated attack of a passivated metal at discrete sites in a corrosive environment. In a metal, it may initiate because of (i) a breakdown of the otherwise protective passive film and/or (ii) the presence of metal surface heterogeneities, such as grain boundaries or inclusions. Localised corrosion poses challenges for detection, and once initiated, the damage propagates rapidly, which results in unanticipated and sometimes catastrophic failures of the materials. 1 Localised corrosion may induce significant repair, maintenance and replacement costs of equipment, transport vessels or buildings. 2 Therefore, a successful development of purpose-designed corrosion-resistant alloys and inhibitors may have significant commercial benefits.
It is conceivable that future materials will be engineered from individual atoms or molecules, and researchers will be able to design molecular configurations and processing routes to obtain the required performance. 3 Key elements of a material are being designed on the molecular level (e.g., inhibitors and their interactions with metal surfaces), and the complexity and dimensions of these deliberately designed molecular elements will presumably increase in the near future. However, molecular design will permit myriad combinations (and permutations) that require validation to determine the most effective combinations. Combinatorial and high-throughput methods 4 are accelerating the pace of material discovery and optimisation. However, although they are faster than the traditional methods, these techniques are not sufficiently rapid and typically generate data only in the laboratory with no clear method of linking to the in-service performance. Combining computational modelling with high-throughput methods can not only significantly increase the speed of material discovery but also provide a link to the in-service performance. Using computational design, designers will be able to access realistic predictions of the properties and the performance based on the actual microstructure and the operating environment. In this regard, computational modelling may be considered a vertex of a triangle, where the other two vertices are represented by experimentation and the formulation of theories. 5 This comprehensive approach is especially beneficial in studying localised corrosion, where the inherently low rates of material removal make experimentation a relatively lengthy process, even under accelerated conditions.
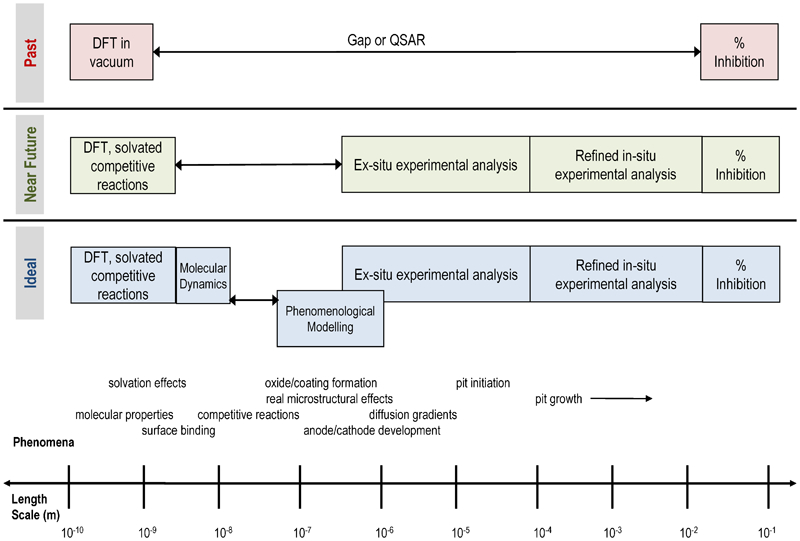
Localised corrosion is inherently multiscale (see Fig. 1) because the nucleation and the propagation of pits are affected by phenomena on vastly different length and time scales (e.g., the surface interactions at the atomic scale v. the environmental conditions that are described in the continuum scale). Thus, the corresponding computational design must be based on a multiscale approach. Figure 1 illustrates the length scales of local corrosion from environmental conditions in the continuum, i.e., the macroscopic length scale, (e.g., Ref. 6) to the mesoscopic scale that defines the material microstructure and ultimately to the atomic scale in charge transport phenomena (e.g., Ref. 7). Despite this complexity, as a design tool, an effective multiscale model (MSM) 5,8–11 must seamlessly combine the continuum and the atomistic descriptions of matter. 12 Future design 13,14 at the molecular level will require this type of MSM. From this perspective, in the current work, the literature on such topics as the theoretical modelling of localised corrosion, the numerical techniques, the experimental knowledge required for model inputs and validation, and the limitations in software are critically reviewed. Several thoughts that are relevant to the development of a suitable framework for an MSM are also provided, and the challenges in the development task are highlighted. To the best of the present authors’ knowledge, there is currently no comprehensive MSM in the public domain that simulates localised corrosion in metals from the atomic scale to the continuum scale, although an example of a prospective type has recently been stated. 13 However, there are MSMs for glass 11 and carbon 8 corrosion.
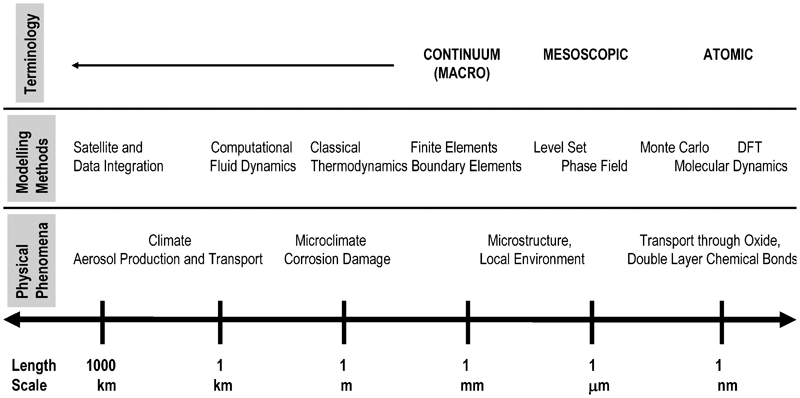
Spectrum of models to describe the electrochemical phenomena at various length scales (approximated)
New paradigms for predictions
A prospective MSM on localised corrosion should provide more accurate and service-relevant predictions than the traditional approaches such as (i) the E-pH or Pourbaix diagrams (thermodynamics), (ii) the polarisation or Evans diagrams (kinetics) and (iii) the Nernst–Planck equation and the transport models that are based on the concentration solution theory. For example, the E-pH diagrams do not account for: (a) the transient behaviour (because they describe only the equilibrium states for given concentrations), (b) the localised variations in conditions such as concentration gradients, and (c) the highly influential features such as the alloy microstructure. Essentially, this thermodynamics-based empirical tool cannot be relied upon to predict the corrosion rates or the degree of passivity or for use in a non-equilibrium situation, although some useful extensions have been made 15 since its introduction. In addition, most Pourbaix diagrams only address pure metals and not alloys (an exception is Fe–Cr–Ni 16 ). The polarisation diagrams do not consider mass transport limitations, which are important when there are concentration gradients in the electrolyte. In addition, different half-reactions (i.e., oxidation/reduction) are activated at different potentials, and they depend on the microstructural features of the electrode and the microenvironment in the electrolyte; thus, several permutations and combinations of these polarisation curves are required to adequately describe an engineering system. Furthermore, the transport models heavily depend on the approximation of macro-homogeneity and local electro-neutrality. Thus, the transport models neglect the atomistic details of the reactions and the inhibition mechanisms that occur on the metal or oxide surfaces. In addition, a common drawback of the above schemes is that they provide information at only one level. An ideal MSM should include strategies to overcome such limitations and the lack of resolution that accompanies the traditional techniques. For example, the alloy microstructure should be sufficiently described in terms of the metallurgical phases, their relative amounts, and their spatial distribution. However, to develop an MSM that provides a major advancement over traditional methods in the medium term, presently unavailable experimental data at the lower scales must be generated to describe the localised corrosion at an atomic level. Traditional electrochemical, surface analytical and spectroscopic studies only provide integral information on the electrochemical processes that occur at the solid/liquid interfaces, but they do not provide information on local atomistic events and the influence of surface imperfections on the interfacial processes. 12 In addition to such considerations, the MSM should seamlessly link the scales and be as computationally efficient as possible. In a recent review of MSMs in materials science, Elliot 5 noted that there are some promising strategies that are making progress at reducing the computational burden of models, in particular, at the lower scales. Thus, it is likely that a full-scale MSM can be solved within reasonable time frames in the not-so-distant future.
Types of existing models
Single-scale models
Based on the assumptions about the behaviour of events (i.e., deterministic v. stochastic) and the scales involved (atomic v. continuum), the available single-scale models may be divided into various categories (Table 1).
Types of single-scale models that relate to localised corrosion
Currently, the deterministic models generally focus on the growth of a single, previously established pit (or crevice) or a collection of pits with predetermined anodic and cathodic sites. However, these models do not incorporate pit initiation events because the currently available experimental knowledge is insufficient to deterministically model the pit initiation, which is an atomic scale event that is difficult to observe physically. Reigada et al. 32 note that this incomplete knowledge has led modellers to consider pit initiation as a sporadic and stochastic event, whose random nature manifests itself in both the distribution of induction time and the amount of current at a constant applied potential. Furthermore, the computationally intensive molecular dynamics (MD) methods such as the kinetic Monte Carlo (kMC) method are required to simulate atomistic events over distances that are relevant to pitting. Therefore, the extents of the systems that may be covered in those simulations are greatly limited, which reduces the practical value of such efforts, unless a multiscale approach was employed. Regardless, given the expanding body of evidence that pertains to the preferential initiation of pits at the microstructural features such as inclusions (e.g., Ref. 49) and second-phase particles (e.g., Ref. 50), it is increasingly likely that atomic scale deterministic models will be developed in the coming years.
Although the stochastic models are elegant tools to model the mechanisms that are not fully understood, ipso facto, they cannot be interrogated to gain a deeper understanding of the influences that are exerted by causal factors. Thus, these models lack the resolution to define a process at the level of detail that is necessary to design corrosion-resistant systems. In addition, the stochastic models that the present authors found were developed in the macroscopic scale, except for the Monte Carlo (MC) model by Reigada et al., 32 which solved both the electrochemical responses and the morphological features by assuming that the pit propagation is a tunnelling process.
The unified models (e.g., Refs. 46–48) have sought to combine the elements of the deterministic models and those of the statistical models to redress the limitations of the two approaches. For example, Laycock et al. 47 developed an experimentally validated hybrid model, where a purely stochastic model for pit initiation was combined with a deterministic model to propagate single pits in stainless steel (SS). Such an approach is perhaps most suitable for the early versions of the proposed MSM.
Linking of scales
An MSM may speed up computations by replacing the atomistic models with a less computationally demanding continuum assumption at locations that are removed from the region of interest. In addition, it extends material behaviour to the larger scales that represent the continuum realm. Thus, an MSM makes predictions relevant to the engineers.
Ingram 9 has discussed different frameworks that may be adopted to link the scales (Fig. 2). This figure shows how the two-scale domains may be linked in terms of the balance volumes (BVs) using six different frameworks. The broken lines show the regions where such BVs overlap. The BVs of the mesoscopic- and macroscopic- (continuum-) scale submodels are represented by Σμ and Σ M, respectively. In a multidomain model (Fig. 2a ), the BVs occupy the adjacent, non-overlapping parts of a system domain. In some cases, there is a small interface region between the domains where both models apply (Fig. 2b ). In an embedded framework (Fig. 2c ), Σ M spans the system domain, whereas Σμ describes only a portion of that domain. In the parallel framework, both models, which are labelled 1 and 2 in Fig. 2d , span the system domain. In the serial framework (Fig. 2e ), Σ μ does not exist because the associated conservation equations are transformed into constitutive relationships. Finally, in the simultaneous framework, the whole system domain is described by Σ μ . No BV is associated with Σ M equations, since no conservation relations are used at this scale. More details on these frameworks, including how information is transferred between BVs, are provided by Ingram. 9
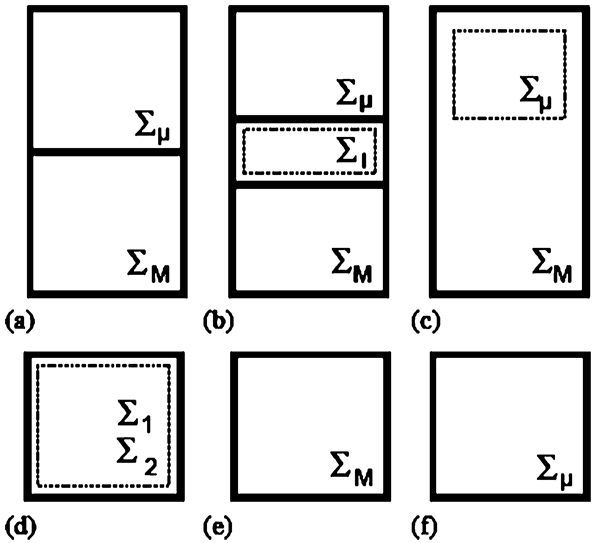
Framework class definitions 9 for the two-scale models: a multidomain, b multidomain (with interface zone), c embedded, d parallel, e serial, and f simultaneous
To circumvent the large computational load at the lower scales, the multiscale approach adopts coarse graining towards the higher scales. For example, in an embedded MSM, 9,51 an atomistic model may be embedded within a mesoscopic model, which can be embedded within a continuum scale model. Then, the lower-scale model can update the higher-scale model at regular time intervals with the corrosion damage information (e.g., Ref. 52, the simulating fracture). Moreover, one should also consider the potential application of meshless methods 53–56 to pit propagation because these state of the art techniques are being used increasingly successfully to simulate crack propagation (e.g., Refs. 53 and 54) in the field of computational material science. These methods are better suited to tackle the moving discontinuities such as crack propagation along arbitrary and complex paths, whereas the traditional finite element method (FEM) methods would involve considerable meshing and re-meshing. There are significant inherent challenges in attempting to link the scales, and these challenges are well understood. 5,12 Elliot 5 and Tan 12 discussed several numerical methods that have been used for such linking at the boundaries of the two domains. In the Numerical strategies section, the present authors discuss the numerical difficulties that must be surmounted before a comprehensive MSM for localised corrosion may be successfully built.
Formulation of crucial phenomena
This section discusses the barriers and the data requirements for MSM, namely, the crucial parameters and events 57,58 that influence the localised corrosion. This discussion on formulation ends with a pictorial summary (see Fig. 5) of the types of models that are currently used.
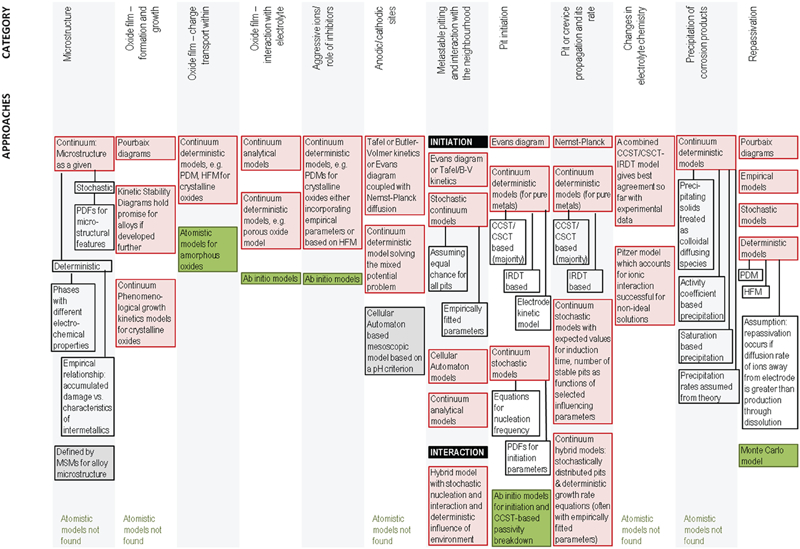
Summary of approaches found in literature for various events relevant to localised corrosion: continuum approaches are shaded in red and atomistic in green
Composition of the metal substrate: its formation and influence
The microstructure of a metal or an alloy and its processing route control the sites of inclusions, the second-phase particles, the solute segregated grain boundaries, the flaws, the dislocations and the degree of surface roughness, all of which overwhelmingly influence the preferred locations for pit initiation. These quantities are discussed in detail elsewhere. 15,50,59–61
The present review suggests that the existing continuum models are too limiting in their treatment of the microstructure. However, there are early MSMs that may be used as starting points to build future MSMs that comprise microstructure evolution during solidification and link a microstructure to in-service performance.
Continuum models: rudimentary description of microstructures
Several early single-scale (continuum) corrosion models (Sharland 62 ) and many recent works (e.g., Refs. 29, 63 and 64) did not consider the microstructure. However, the modellers who did, assumed the microstructure as a given. The modellers who followed the deterministic path (e.g., Refs. 65–67) have defined the microstructure either in their computational grids 65,66 or treated them as a regular array and used analytical solution methods. 67 The modellers who work in the stochastic realm have used probability density functions (PDFs) (e.g., Ref. 68) to account for the probabilistic nature of the features. Brown and Barnard 65,66 incorporated the microstructure that defines a certain electrochemical property (e.g., different Tafel dissolution kinetics for individual phases) for each finite difference method (FDM) computational cell. Their latter model 65 could predict the distribution of cathodic regions. Jakab et al. 67 also developed a simplified deterministic model by treating a heterogeneous AA2024-T3 electrode as a regular array of Cu-rich favoured cathodic sites (partially covered with an inactive aluminium oxide layer) in a benign Al matrix. Furthermore, Zhang et al. 68 developed a stochastic model for the same alloy AA2024-T3, which, according to the authors, provided a new approach to the prediction and quantification of localised corrosion kinetics based on the alloy microstructure. Their PDFs for grain dimensions had parameters that were fitted based on the observed three-dimensional grain sizes, and a ‘brick wall model’ was used to model the grains (and the inter-granular regions) in 3D. There are also empirical models that correlate the corrosion consequences with the microstructural aspects. For example, Cavanaugh et al. 69 developed an empirical relationship between the accumulated corrosion damage in AA7075-T651 and the physical and electrochemical characteristics of the intermetallic particles, and this model predicted the pit radii (assuming hemispherical pitting) as a function of the immersion time in the 0⋅1 M NaCl. However, such models have limited applicability and cannot be relied upon for any experimental conditions other than those for which they were developed. In summary, the traditional continuum models that incorporate the microstructure have assumed it as a given quantity and have described it in relatively simplistic terms. No work appears to have considered details such as the recently observed 70 non-random clustering of buried intermetallic particles in AA2024.
Multiscale models for solidification and prediction of service life: early versions are promising
Microstructure evolution during solidification
The formation of microstructure originates at the atomic scale, where the initial nucleation and the growth of critical nuclei occur. 10 This formation is followed by the growth of microstructure at the intermediate mesoscopic scale. Rafii-Tabar and Chirazi 10 have reviewed several deterministic, stochastic, hybrid models and some early MSMs that predict the microstructure evolution; they have also developed a well-documented, validated, generic MSM that spanned the nano-meso-continuum scales for microstructure formation (Fig. 3).
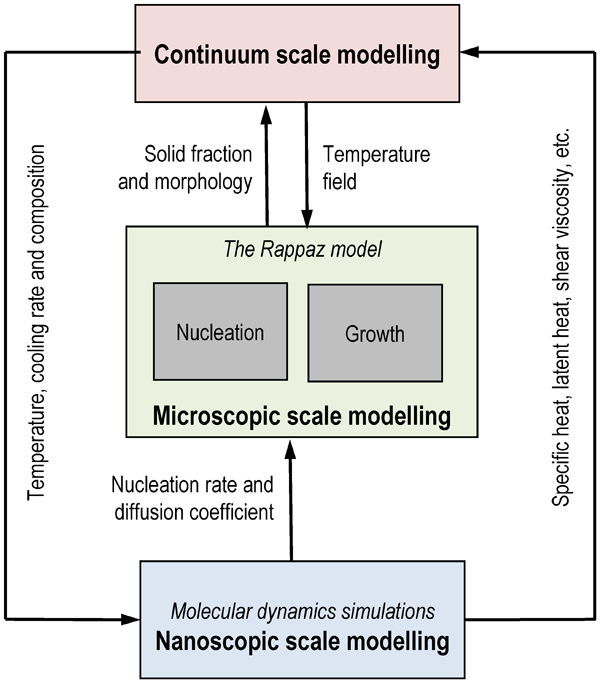
Multiscale model (MSM) for solidification by Rafii-Tabar and Cherazi 10
Their MSM is representative of the limited number of available MSMs for microstructure prediction that can be linked to an MSM for localised corrosion. In the Rafii-Tabar and Chirazi model (Fig. 3), MD simulation techniques were used at the atomic scale and linked to a cellular automaton (CA)-based mesoscopic model for microstructures. The Rappaz model 71 that was used for the microstructure combined a stochastic approach to the nucleation of grains (which was implemented using CA) with a deterministic, diffusion-controlled approach to their growth. The parameters that were used in the Rappaz model were calculated using MD simulations. Finally, the continuum scale simulations were performed based on finite element (FE) and finite volume (FV) techniques. The models at different scales were executed independently and coupled through databases using a serial framework (Fig. 2e ). The procedure was to associate an MD simulation box with a CA volume to couple the nano-meso models, followed by associating a CA volume with a finite volume to couple the meso-macro models. Because only one-way coupling is possible in a serial framework, and because the three scale models were run separately using different codes, it was necessary to run the meso and macro models more than once to achieve the two-way coupling between the scales through iteration. For example, in its first run, the meso model generated the inputs for the macro model, and in the second run, which was performed after the macro model had predicted the temperature evolutions, it predicted the microstructure-based on the temperature histories and the cooling rates.
Influence of microstructure on in-service performance
The MSMs to link microstructure with the in-service performance at continuum scales for steel were published by research teams that were led by Olson. 72,73 From these MSMs, some concepts and strategies could be borrowed to incorporate the influence of microstructure on the corrosion performance at higher scales. The Olson models link the microstructure of steels with failure scenarios such as ductile fracture by predicting the relevant properties. The microstructure was decomposed into multiple scales using a nested domain framework (i.e., an extension of the embedded framework, Fig. 2c ). The continuum scale was linked to an embedded microscopic scale (primary inclusion particles) and again to another embedded sub-microscopic scale (secondary inclusion particles). Thus, their FE solutions could account for the influence of inclusions during fracture in terms of the particle size and the carbide/matrix debonding stress.
Recent advances
As the Rafii-Tabar and Chiraz model 10 applies in its current form only to binary alloys, the important effect of the alloying elements on the corrosion morphology 50 cannot yet be modelled. Therefore, the more recently developed and validated MSM models 74–76 are more attractive for the corrosion modellers who work with engineering alloys, although these models span only the meso to continuum length scales and the associated temporal scales. These latter models are useful because they are capable of handling both ternary 74,76 and quaternary 75 alloys that are industrially more relevant. These 3D models can also account for the influence and the interaction of multiple phases (solid/liquid/gas) during the microstructure development. In addition, some of the latter models (e.g., Wang et al. 75 ) have further enhanced their realistic nature by accounting for the effects of the cooling rates and the alloy content in their prediction of the microstructural features that included the shape, the size and the distribution of the intermetallics and the defects such as porosity. The main output of these models is typically a suggested deterministic microstructure. The information flow between the two levels in the Wang et al. 75 model was facilitated through the coupling of temperature and pressure variables (see Ref. 76). In addition, the lower-scale model was implemented as a subroutine of the macro model, which was solved using FEM in an embedded multiscale framework (Fig. 2). The mesoscale sub-region formed a total volume fraction of approximately 0⋅001% of the domain volume that was covered by the continuum scale model, which allowed for the mesh sizes in the meso model to be on the order of micro metres. In summary, the microstructure-evolution related MSMs treated the nucleation events stochastically (e.g., with a pre-set nuclei density and a nucleation potential with a Gaussian distribution PDF 75 ) and modelled the growth events deterministically using a hybrid strategy. Thus, there is no purely deterministic microstructural model. Regarding MSMs for linking the microstructure to the prediction of service life, validated works 72,73 by Olson’s teams appear to be the most pre-eminent, and their strategies may be used as a basis to link the microstructure to the in-service corrosion performance in MSMs.
Proposed improvements
Because the existing models on corrosion treat microstructure as a given quantity or have a simplistic description of microstructure, the ability of an MSM (that incorporate such models) to tailor materials is limited. An MSM on microstructure prediction should ideally be linked to an MSM on localised corrosion, so that through a feedback loop, the alloy system may be optimised for corrosion performance. Furthermore, none of the existing models account for the formation of the oxide layer (the Passivating oxide film, its thickness, and its porous nature section below), and they do not provide other corrosion-related information such as the interfacial energies at the grain boundaries and the grain mismatch, which are indicators 77 of the propensity of various sites to corrode in some cases. Therefore, in the long term, it is desirable to have the microstructure model also predict the composition and the morphology of the oxide film. Recently, promising efforts at atomistically simulating the precipitation kinetics in the solidification of multicomponent interstitial/substitutional alloys 78 have been published. It is also desirable to predict the surface roughness characteristics. However, in the short term, the prescription of the entire final microstructure as a given quantity remains a convenient starting point.
Passivating oxide film, its thickness and its porous nature
The phases that make up an oxide film that forms on the surface of a substrate are often traditionally identified using Pourbaix diagrams, 59 although these diagrams only describe the equilibrium phase distributions. The passivating nature of the film is attributable to its limiting influence on charge transport between the electrolyte and the substrate. The oxide film thickness varies between nanometres (e.g., Refs. 79 and 80) and single-digit micrometres (e.g., Ref. 81). Different oxides have different degrees of stability depending on the environmental parameters 82 such as the pH 81 , the corrosion product concentration and the ionic species. Almost all passive films have multilayer structures, usually with the inner oxide and the outer hydroxide parts, the former is the barrier layer against cation transfer, and the latter is an exchange layer with the electrolyte. 83 A review 7 on passive films at the nanoscale has provided rare insights into the structure and the growth of oxide films based on recently observed atomistic-level details. In addition, the passivating films of Zn and Fe are crystalline oxide grains with grain boundaries, whereas those of Al alloys and SSs are amorphous in structure. 84 The electrochemical behaviour of the oxide film, its thickness and its chemical composition depend on numerous parameters. 79,85–88 Because this oxide film is somewhat analogous to the artificial coatings 86,89,90 that are designed to decelerate corrosion, some comments in this section are equally applicable to those coatings. The Pourbaix diagrams cannot be accurately applied to passive films that are non-equilibrium structures, the existence of which depends upon an appropriate relationship between the rate of formation and the rate of destruction. 91 The recently introduced kinetic stability diagrams (KSDs) 91,92 may provide a means to calculate the corrosion rates for alloys if they are further developed; however, their construction is tedious because there are numerous possible combinations of electrode potentials, ionic concentrations and pH.
The present review notes that the continuum models do not have sufficient resolution to describe the atomistic mechanisms, and a lack of experimental data at lower scales makes it more difficult for atomistic models to be developed in the short term to describe the behaviour of oxide films.
Continuum models: lack resolution
Transport through oxide film
Crystalline oxides: Several continuum phenomenological growth kinetic models have been proposed to model the growth of oxides and the rate-limiting cation-transport mechanism through the oxide thickness. Many of these models have been reviewed by Hendy et al. 21 All of these models assume the homogeneity of the crystalline oxide to which they are applicable and have several free parameters that must be fitted empirically. Hendy et al. 21 showed by comparing their own ab initio simulations with those of the phenomenological models, that the latter models were consistent with the calculated barrier energies only if it was assumed that the grain boundary diffusion of cations dominated. These results are consistent with the sentiments of Marcus et al., 28 who proposed that the cations would preferentially migrate through the more electrically conducting inter-granular regions. These findings highlight the importance of considering the grain boundary structure and their inter-connectivity in the modelling efforts.
Amorphous oxides: For these oxides, models that relate to ionic conduction have been relatively scarce. In a model that was proposed by Wang and Hebert, 93 the current was carried by defect clusters, which were created by the inward displacement of O2− ions around an O vacancy in response to the vacancy’s electric field. The model hypothesised that the displacement created a gap between the first layer and the second layer of O2− ions that surrounded the vacancy, within which the metal ions could easily migrate with little required activation energy. The analytically solved steady-state equations were satisfactorily validated using experimental results.
Interactions between oxide film and electrolyte
The interactions between the oxide and the electrolyte determine the rate of electron transfer presumably by altering the electronic work function at the interface (e.g., Vago et al. 94 ). However, although phenomena such as O2 reduction, oxide transformations and electron transfers to redox species in a solution occur at an atomic scale, it would seem that atomic scale models are not yet available in the public domain. Only continuum scale analytical models by Jakab et al. 67 and Chen et al. 95 appear to address this phenomenon. In some cases such as the ZnO system, the ability of oxides to act as reducing sites can significantly change the nature of the cathode and reduce the diffusion limits. 96 For example, when pH<5, the cathodic reactions in the Zn system are rate-determining. 61 In such cases, the model must account for the scenario where the porous layer on the metal surface either competes with or complements the oxygen reduction reaction (ORR) that occurs on the metal surface. This scenario can be affected by many factors, and a recent continuum scale porous-electrode model 97 has incorporated some of those influences. Nevertheless, an atomistic-level deterministic treatment of the interactions is preferred because the mechanisms at play operate at that level.
Although the metal surface can ideally be represented using a crystalline lattice, the oxide layer may or may not be crystalline depending on the metal and the conditions under which the oxide is formed. Most amorphous solids are initially hard, deform notably little, reach their plastic limit early and break because they have high strength and low ductility 98 . Multiscale modelling of amorphous solids is far less advanced than in crystals because of the limited understanding of the behaviour of plasticity in amorphous materials. The integrity of the oxide layer on an active metal is highly influenced by the stresses that the metal beneath experiences. These stresses can locally rupture the oxide layer, which leads to cracks (also known as holidays), through which the corrosive solution can percolate and reach the surface of the metal. In addition, when the oxide layers host a cathodic oxygen reduction, the net corrosion rate becomes vastly different from a situation where the oxide layers do not host such an electrochemical reaction. Finite element method could ideally handle this situation in the continuum description. Note that unless the oxide layer is electrochemically active, we may not be required to model the entire oxide layer in a multiscale manner even if the oxide breaks down locally.
Atomistic models and paucity of data at the atomic scale
The continuum assumption of a uniform passivating layer for the complex heterogeneous structure of the oxide film is quite inadequate. In addition, as noted by Williams et al., 99 although the continuum models for migration and accumulation of point defects in the film are appropriate to describe the behaviour of high-purity single-phase metals, they are inefficient for the engineering alloys. Thus, atomic scale models are required. Maurice and Marcus 7 reviewed some of the atomistic models and highlighted their usefulness in helping one understand the mechanisms that occur at the lower scales. The ab initio model of Bouzoubaa et al. 18 predicted that the surface undulations on the oxide film played a major role in its chloride thinning mechanism. Kim et al. 100 used DFT to understand the origins of the natively n-type characteristics of ZnO by modelling the interaction of a Zn interstitial with an oxygen vacancy. The 3D atomistic model of Diawara et al. 101 successfully modelled the selective dissolution and the passivation of Fe–Cr alloys by simulating the formation of oxide nuclei from chromium-rich clusters on the surface. This model confirmed the experimental observation that Cr preferentially diffused towards the Cr clusters on the surface, whereas iron atoms showed no such preferential diffusion. Their kMC model also showed that passivation occurred within a matter of seconds. The ab initio modelling work by Costa et al. 20 on SSs studied the effect of water coverages in the Cr2O3 film on the surface, and the energies of adsorption that it predicted were successfully validated with experimental data. Similarly, Hendy et al. 21 calculated the energy barriers that were associated with the transport of cations through the oxide film by combining ab initio simulations with experimental observations at the atomic scale. These investigations reinforce the value in modelling the film at the atomic scale. However, in this connection, Maurice and Marcus 7 noted that the development of such models will be limited by the complexity involved in considering the three phases (alloy substrate/oxide/electrolyte), their interfaces, the electric field, the temperature, the orientation of the oxide film, its nanostructural defects and the surface defects. The issue of heterogeneous passive films on engineering alloys makes it critically important to select what is modelled. The examples above are limited in terms of computational load when attempting to model engineering alloys even with a reasonably low concentration (in relation to the number of atoms that can be considered in the ab initio methods) of second-phase particles. This problem is even more challenging when pit initiation is considered. Current ab initio models are rather weak at modelling the defects that exist at second-phase particles in a matrix because the required number of atoms is too high.
The relative scarcity of models at the atomic scale is unsurprising given the computational burden that we discussed above and because the experimentalists could not observe the oxide film in detail at an atomistic level 28,79 until recently. Nevertheless, there is an increasing amount of literature (e.g., Refs. 79, 99, 102 and 103) that addresses such measurements, slowly filling the present gap in knowledge at such length scales.
Proposed improvements
The oxide film is a classic example of the need for a multiscale approach to corrosion modelling because its atomic scale thickness is coupled with its mesoscopic-scale microstructure, which is punctuated with continuum scale defects. Thus, either a three-scale approach or a less comprehensive two-scale platform (meso/continuum scales) may be chosen to model the film. However, in the latter approach, a generic phenomenological model that adequately describes, in the meso or continuum scales, those mechanisms 104,105 that actually occur in the nanoscale through derived transport and kinetic parameters will be considered mandatory. The advanced version of a prospective MSM should have the capacity to predict the oxide composition and structure (both geometric and electronic), including its thickness and its porosity distribution, based on a given microstructure of the substrate and the electrochemical environment. Of course, this hypothesis assumes that sufficient experimental data become available for validation in the short to medium term. For a deterministic model to have a strong predictive capability in relation to the electrochemical behaviour of the oxide film, the following must be undertaken: 106 (a) the kinetic and the transport parameters that are pertinent to different metallic constituents of the film must be determined either based on the available values or using advanced experimental techniques, and (b) the energy heterogeneity of the transport medium must be considered using the distribution functions for the diffusion coefficients of individual species in the compact film and by quantifying the role of the grain boundaries in the transfer of matter and charge through such a film. The model should also account for the interaction between the film and the environment using kinetic data (and thermodynamic data) on the adsorption, the surface complex formation and the re-precipitation reactions of various cations at different pH levels and locations of the oxide. It is also desirable that the atomistic modelling of nano-scale events such as the oxide penetration/thinning mechanism (e.g., Refs. 18 and 28), which leads to the prediction of nucleation sites. In addition, some of the free parameters required for the phenomenological models may be determined from first principles through such simulations where theories are available, rather than being fitted empirically. However, given the enormous effort and time required to achieve all of the above, the early versions of an MSM are likely to incorporate approximations for many of the aforementioned aspects.
Influence of aggressive ions (pH and Cl−) and role of inhibitors
The acidity or the alkalinity of the environment significantly affects the corrosion behaviour of metals, and there are several rules and exceptions that govern such behaviour. 50,57,59,61,107 Traditionally, the influence of pH on the corrosion tendency of metals in aqueous solutions of a given concentration has been described using Pourbaix diagrams, the limitations of which have been mentioned in the New paradigms for predictions section (see also Ref. 92). The inhibitor molecules in the electrolyte also significantly influence the corrosion rates, but unlike Cl− ions, they slow down corrosion. Inhibitors are an attractive option when thick coatings are either unacceptable for isolating the metal from the electrolyte or when coatings may become damaged. In addition, inhibitors work by adsorbing onto the metal surface at its interface with the electrolyte 108 and creating nanometre-thick films instead of the usually millimetre-thick coating layers.
The current review found that the continuum scale models could not be used in a generic MSM because of their lack of generality, and the rapidly improving suites of atomistic modelling software packages are increasingly facilitating the modelling in this area at lower scales.
Continuum models: lack of universal applicability
Traditional models 25,47,62,109–114 have either neglected the influence of pH or Cl− or incorporated their influence in a rudimentary manner. Sharland 62 has reviewed some of these models. More recently, as noted by Frankel and Sridhar, 115 a point defect model (PDM) 116 incorporating empirically determined parameters correctly predicted the logarithmic dependency of pit initiation on Cl− concentration, although it could not calculate the often observed bi-logarithmic dependence. The Cl− influence was integrated in the PDM by hypothesising that its concentration altered the generation and the transport of cation vacancies. A simplified PDM for iron, 117 which was based on the high field model (HFM) formulation, also predicted the correct qualitative trend for the dependence of the film thickness on the pH of the electrolyte. Nevertheless, neither PDM nor HFM can be universally applied (they are only valid for crystalline materials) and are limited by the assumptions (e.g., homogeneous oxide) and the need to fit the parameters empirically for each scenario. Lastly, the effect of the inhibitors has not been tackled by continuum models as much as the present authors are aware, which is understandable given the patent lack of resolution of such models in accounting for the surface/liquid interactions at the molecular level. In summary, traditional approaches such as Pourbaix diagrams are inadequate, and the continuum models lack universal applicability and resolution in describing non-homogeneous microstructure. Thus, although some traditional models may have to be used in early versions of an MSM, a truly generic advanced MSM will require more detailed modelling at the lower scales.
Atomistic models: promising, but currently at early stages
The atomistic models of more recent origin have shown promise in this area by incorporating the influence of ions in the aqueous solution from a more fundamental point of view. Molecular or atomistic modelling brings to multiscale modelling a unique tool that can discriminate behaviour on the molecular scale 118,119 and allow a molecular design. Consider the development of organic corrosion inhibitors. Their performance is strongly controlled by bonding (electron sharing) and the type of pendant functional groups that are attached to aromatic rings. However, the exact molecular structure has a critical impact on the inhibition performance; because conventionally, inhibition is measured by exposure of metal plates in inhibited solution or by electrochemical tests, a direct correlation to the inhibitor’s efficiency does not exist because of the multiple scales that are involved. Hence, incorporating molecular modelling into a multiscale modelling framework offers the possibility of forming a continuous link from the molecular structure to the inhibition of anodic and cathodic activity on the surface, which inhibits corrosion.
Density functional theory methods
A detailed account of the evolution of how the Schrodinger’s equation is solved in DFT can be found in existing literature, e.g., Ref. 120. In the modelling of surface/molecular interactions (such as inhibitors), the most commonly used basis sets are plane waves (see Table 2). The localised basis sets first approach (e.g., in Spanish initiative for electronic simulations with thousands of atoms, SIESTA) the edge plane waves (e.g., in Quantum ESPRESSO) because of the lower computational requirements that are associated with modelling the vacuum space.
Density functional theory (DFT) software packages
LSDA: Local Spin Density Approximation.
GGA: generalised gradient approximation.
LCAO: localised combination of atomic orbitals.
Taylor 121 has reviewed some works in this space. Bouzoubaa et al. 18 modelled the aggressive role of Cl− in breaking down the passivity of the crystalline NiO using a periodic DFT. They intended to investigate the interaction of these ions with a stepped surface on the hydroxylated NiO film, which is characteristic of the barrier oxide layer on passivated nickel. The results suggested that adsorption of Cl by exchange with surface hydroxyls is energetically favourable but may not promote dissolution. Sub-surface insertion into the lattice was found to promote dissolution only if the Cl surface coverage was>70%, which is unlikely because of the Cl–Cl repulsions. Thus, this work, which was performed on a defect-free surface, did not confirm the existing hypotheses of adsorption-induced surface thinning or Cl sub-surface penetration. Bouzoubaa et al. later extended their work to include various halides. 17 Although a final word on the hypotheses cannot be provided until a more realistic surface with defects is simulated, the above works display the power of atomistic simulations besides putting conventional wisdom to the test.
Inhibitor/surface modelling
Inhibitors also have been modelled at the atomic scale by several works (e.g., Refs. 122–129), and the literature contains a wide range of approaches to model their interactions and electrochemical effects, some of which were reviewed by Gece. 122 In addition, although several software codes such as Vienna ab initio simulation (VASP), Gaussian and Quantum ESPRESSO have been used, a relatively recent entrant known as SIESTA 128 appears promising in the area of inhibitors because its underlying method can describe the interaction event well. Much of the current work models the structures of inhibitors in vacuum 130,131 and obtains the correlations 132,133 with percent inhibition for a metal in an inhibited saline solution. However, more recent studies 134 include solvation effects, examine an inhibitor’s binding to the surface, consider the charge double layer 135 and model the actual kinetics of the cathodic reaction. 124,136 The effect of solvation is considered by placing water molecules in vacuum. However, two issues must be overcome: 134 (a) the probable water structure must be estimated prior to its placement (otherwise, the DFT code will spend all of its time on optimising the water structure), and (b) the long-range interactions such as hydrogen bonding and dispersion/Van der Waals forces must be included. To include the dispersion forces, it is necessary to add an auxiliary force field to the DFT calculation with a number of approaches that are developed to do so including: (a) the non-local functional (vdW-DF functional) of Langreth and Lundqvist, 137 (b) the modified pseudopotentials (von Lilienfeld, 138 (c) the highly empirical (hybrid) metaGGA functionals, 139 and (d) the interatomic (pairwise or beyond) dispersion corrections such as that of Grimme. 140 The approaches vary widely; for example, Langreth and Lundqvist explicitly incorporated the pairwise point–point interaction while ignoring the non-additive many-body interactions, whereas Grimme’s method is a highly empirical approach based on the parameterisation of the interaction energies.
Modelling the electric double layer
Generally, the double layer effect on metal surfaces is modelled by adding or removing a charge from the unit cell of the metal and balancing with a homogeneously applied counter charge. 140 This approach can be problematic in plane-wave methods such as those used in VASP because the charge may not be well localised. Spohr 141 has reported an increase in the use of ab initio models to simulate the interfacial interactions between the electrolyte and the solid substrate by modelling the electric double layer. One such work is by Taylor et al., 142 who examined the double layer regions for water over a range of metals and compared the equilibrium potentials for the initial steps of water reduction and oxidation at the surface with known experimental quantities. Spohr 141 and Yeh 143 found that the inclusion of an explicit solvation provides more realistic reaction energetics in comparison to the solutions in vacuum. Similarly, in a related work, Janik et al. 124 simulated the electronic double layer by adding various numbers of electrons in a Pt unit cell and adding a compensating background charge. However, Taylor et al. 144 noted that the effect of charge addition highly depends on the orientation of water dipoles of the chosen solvation structure, and the fluctuations of the water molecule orientation can induce instabilities in the charge localisation. This phenomenon allowed the potential at the surface to be controlled by adding fractions of charge. They concluded that: (i) the increasing use of molecular simulations allows the combination of statistical mechanical description of the double layer with a description of elementary chemical processes on the electronic structure level; (ii) the free-energy methods are applied to describe the chemical reactivity within and beyond the framework of the continuum Marcus theory of electron transfer, 145 and (iii) at sufficiently high concentrations, direct simulations of the two-phase systems with an aqueous solution and a charged or uncharged solid phase or surface can model the entire double layer region. Taylor 121 also discussed the specific types of information that may be garnered from DFT simulations of various metal–environment interactions and the associated challenges. To summarise, the atomistic models that address different mechanisms at the electrolyte/electrode interface, including the influence of ions, have begun to appear in the open domain and have provided useful insights into the interfacial dynamics. In the large domain of inhibitors, although significant progress is being made, major challenges must be resolved, including how to effectively model the solvation, the potential and the chemical reactions at the double layer of the metal/solution interface.
Bridging the nano-gap
The design of inhibitors must span the scales from 10−10 to 10−1 m (the scale of a test plate) and cover a wide range of phenomena (see Fig. 4). The properties derived from DFT in vacuum studies may not be relevant to the inhibitor surface-binding and the link between the molecular properties and the mesoscale phenomena such as anode/cathode development, pit initiation, and pit growth is not evident. Thus, a large gap currently exists between the DFT models and the measured inhibitor efficiency in terms of both scale and phenomena. This gap is often 133 covered using a pattern recognition or a neural network approach such as the quantitative structure activity relationships (QSAR). In the near future, the DFT models will likely accurately include the solvation effects and model the competitive reactions (i.e., oxygen reduction v. inhibitor absorption), and local electrochemical techniques such as local EIS can be used to develop refined local parameters and their spatial variation (e.g., coating resistance or diffusional properties), whereas the post-test analytical procedures can examine the structure of the protective layers on the metal surface (i.e., using X-ray photoelectron spectroscopy (XPS), etc.) down to the sub-micron scale. Two additional developments can further close this gap. The DFT studies can be linked to MD studies, which will allow an order-of-magnitude expansion on the molecular scale. 125 Lastly, most electrochemical formulations are based on the Butler–Volmer equation, which provides an average free-energy formulation of charge transfer but does not examine the individual processes that are involved in such transfer. Recent phenomenological models 146 break down the charge transfer in the solution into a number of components and allow each component to be addressed, which effectively refines the electrochemical scale.
‘Nano-gap’ between density functional theory (DFT) models and measured inhibitor efficiency
At this point, a brief note about the use of ex situ techniques such as XPS is appropriate. In such techniques, the specimens are removed from the experimental solution and placed in vacuum for examination. Such inspection under vacuum may significantly alter some aspect of the surface; for example, dehydration can occur, and the physio-sorption between the inhibitor and the surface will not be maintained, although chemical bonding and chemo sorption in particular will be. Thus, such techniques must be used with care, but they can provide valuable information.
Proposed improvements
Based on atomic modelling, a more fundamental approach can be used for both crystalline oxides 21 and the amorphous variety 147 and is well suited to describe the solid/oxide bonding 148 and oxide/electrolyte interactions. 17,18,141 This approach is likely the preferred option for a design-optimisation MSM because of the fine resolution that it provides. There is currently no such exhaustive and generic model, and until one becomes available, empirical correlations that are based on mesoscopic and continuum scale models must be used to build an MSM. In the intervening period, however, it may be possible to explore the use of KSDs to model the influence of pH and aggressive ions at the higher mesoscopic and continuum scales. The current DFT work addresses the accuracy of surface-binding studies and can also examine the competition between inhibitor binding and the cathodic reactions. Hence, the objective of correlating DFT or MD 149 studies with a coarse parameter such as the experimental inhibition efficiency may have to be modified in the light of data from electrochemical and analytical techniques such as XPS, which probes the surface. Lastly, most electrochemical kinetic models are based on the Butler–Volmer equation, which provides an average free-energy formulation of charge transfer but does not examine the individual processes that are involved in such transfer. The proposed MSM should also address the commonly used but inaccurate assumption that uniform conditions exist in the bulk solution; for example, this assumption often neglects the concentration gradients that extend into the bulk solution from a pit or a crevice.
Establishment of anodic and cathodic sites
Anodic and cathodic sites are characterised by different potentials because of the variations in chemical composition encountered in the alloy and/or the electrolyte. These two quantities (i.e., potential and composition) are coupled in the electrolyte through the Nernst equation under equilibrium conditions. For a large cathode area with a small anode area, 150 the distance between these sites 151 and the protective films that formed on their surfaces affect the corrosion rate. A small anode area results in the rapid penetration of the pit or the crevice because the current density at the small anode is notably high, and the anodic polarisation in chloride solutions is extremely limited. When the cathode size is limited, 22 the limiting corrosion current is determined by the size of the cathode (e.g., Ref. 152). The establishment of the anodic and cathodic sites is dictated by several factors, which are mentioned elsewhere. 59,61,69,150,153–158
The present review suggested that the continuum scale models lack the necessary resolution to describe anodic and cathodic sites and that the development of more suitable atomistic models will need to be supported by the generation of experimental data at the lower scales.
Continuum models: contain limiting assumptions
The continuum philosophy considers that while the potential difference on the corroding surface is zero, there are anodic partial currents across the metal–electrolyte interface at microscopic surface sites, the total of which is balanced by an equal value of the total cathodic current that is similarly distributed under free corrosion conditions. This balance is achieved at the open circuit potential (OCP) of the ‘homogeneous’ metal substrate, and is determined by the rates of the partial electrochemical reactions 159 (which are described by either the Tafel or the Butler–Volmer equations). This information is sufficient for modelling the O2 reduction-driven cathodic tendency only when the influence of the microscopic inclusions and the alloying elements are ignored. There is a limited number of continuum works (e.g., Ref. 160) that address the anode–cathode separation, and they are rudimentary. A model that automatically predicts the anode–cathode separation by solving the mixed potential problem was developed by Venkatraman et al. 64 for corrosion under a differentially aerated NaCl droplet, which was deposited on a Zn substrate. The surface was assumed to be homogeneous; thus, no microstructural details were considered. The previously visited model of Brown and Barnard 65 could predict the cathodic regions. More recently, researchers have started to address the microstructural inhomogeneity-driven phenomena that are relevant to the anode–cathode separation in alloys, e.g., the previously discussed model of Jakab et al. 67 (the Microstructure of the metal substrate: its formation and influence section). In addition, Alodan 161 simulated O2 reduction using an approximated alloy-like microstructure and employed circular discs for the cathodes (inclusions), which were surrounded by an annulus-like region for the anode (aluminium). Although these latter models represent a step forward in the simulation of engineering alloys, they still suffer from the over-simplification of the microstructure. Vautrin-Ul et al. 31,162 developed a CA-based mesoscopic model with a random walk of H+ and OH− ions, where the spatial separation between the anodic and the cathodic sites could be made smaller than that in the continuum scale; thus, the model could more realistically locate discrete cathodic and anodic sites as interwoven within a propagating pit (i.e., not only on the side walls) based on the solution pH at the mesoscopic vicinity. This model was unlike most continuum models, where the cathodic sites are usually located outside the anodic pit or crevice or simply assumed to be along the side walls of a pit. Although it constituted a step up from the continuum scale models in terms of details, the model still lacked the resolution to handle the microstructural details of engineering alloys and was limited by the approximations that described atomistic phenomena at the mesoscopic scale. Therefore, lower-scale models must be developed for the anode–cathode separation; any continuum treatment that assumes homogeneity in the microstructure is inadequate for design purposes.
Paucity in atomistic models and experimental data
Atomistic models were difficult to find for this topic. However, because characterisation studies (e.g., Ref. 50) have started to generate relevant kinetic data in the form of polarisation diagrams for different alloy phases at the mesoscopic level, such models may soon be developed. An alternative that does not depend on experimental data is an atomistic model that is constructed bottom-up from first principles. No such model appears to exist in the current open domain for metallic corrosion, except a model that is a part of a recent MSM by Yu 11 for glass corrosion. This situation is likely to change with the generation of physico-chemical data at the atomistic level (e.g., energies for surface formation, chemisorption, and adsorption) and the increasing application of high-resolution characterisation techniques for corrosion-related studies (e.g., Refs. 79 and 102). Advancing observation techniques such as the in situ atomic scale studies that were reported by Magnussen et al., 163 where the local removal/addition of atoms at atomic kinks at the steps of Cu crystal surfaces during dissolution in 0⋅01 M HCl were observed and quantified, also serve to help accumulate knowledge at lower length scales. However, until a sufficient amount of such data exists, the microstructure-based empirical rules (e.g., for dissolution rates, such as the combination of a chemical rate law (first-order law based on [H+]) and an electrochemical law (Butler–Volmer equation) as used by Suter et al. 49 for MnS dissolution in NaCl) may be necessary. These rules should also quantify the effects such as the probable catalysis by [Cl−] of the MnS dissolution, which was proposed by Williams et al., 164 and any possible influences of the oxide films.
Proposed improvements
One of the reasons for the lack of atomistic models might be the difficulty inherent in coupling the electron transfer (current flow) between atomistic models of anode and cathode. The problem arises because, at the atomistic level, calculations are typically performed for equilibrium configurations whereas the formation of anodes and cathodes are highly dynamic events. It is suggested therefore that the potential use of fluctuation theorems 165 be considered for simulating the non-equilibrium mechanisms. The central premise of these theorems revolves around comparing the probability of phase-space trajectories of the system with that of the anti-trajectories (one that the system would traverse if it were moving in the negative time direction), thus introducing the concept of time-irreversibility into the continuum processes. Thus a non-reversible, non-equilibrium process such as corrosion may potentially be modelled using fluctuation theorems at the mesoscopic level, facilitating the prediction of the evolution of favourable phase-space trajectories. Fluctuation theorems are discussed in more detail later, in the Candidates for future MSM and mathematical coupling of scales section. Early versions of an MSM are likely to have varying degrees of approximations to simulate the anode–cathode separation. One of the available avenues for the development of such models is perhaps the extension of models like that of Diawara et al., 101 discussed earlier in the Influence of aggressive ions (pH and Cl-51) and role of inhibitors section , and that of Legrand et al. 166 These models simulate the selective dissolution of Fe and the passivation of a cluster of Cr atoms (which simulates the Cr2O3 layer) in Fe–Cr alloys. Another pathway is available from the previously noted work of Yu, 11 who simulated the corrosion of glass. This MSM had an atomistic model that was coupled to an MC simulation of surface phenomena such as hydroxylation with cations, chemisorptions, adsorption and dissolution. Then, the continuum scale phase field simulations were performed using material properties that were fed from separate MD simulations. Although the MSM contained approximations, educated guesses and at times relied upon empirically obtained parameters, its framework provides a potential platform for the development of an MSM for localised corrosion.
Metastable pitting
Metastable pitting is generally accepted as the necessary precursor to a stable pit initiation in metallic systems that include SS, 167 aluminium 88 and cast iron. 168 This stage is characterised by the consecutive formation and repassivation of sub-micrometre sized pits below the OCP of an alloy, which leads to oscillations in the potential transients in the active direction in an open circuit during the incubation period for stable pitting. 57,88 Some of the metastable pits survive beyond their usual lifetime of the order of seconds 57 and grow to transition into stable pits under favourable circumstances, which are mentioned elsewhere. 25,50,88,167 Recently, additional causes such as clustering have been suggested to encourage the transition. 169–171 The phenomenon has attracted several theories, including those reviewed by Frankel. 172 In short, from the works of Burstein et al. 173 and those of others (e.g., see Frankel’s comments on Sand’s equation), it would appear that the metastable pits thrive as long as a critical solution chemistry is maintained using various means 173,174 . However, they would die through repassivation following a change to this chemistry, which is precipitated by a catastrophic event such as the loss of the cover at the pit mouth or a violent rupture of the passive film covers, leading to a mixing with the bulk solution. Experimental scientists who work with alloys have reported other conditions that can lead to metastable pits (see Refs. 50 and 175–179).
The present review did not find any atomistic models, although pit nucleation is essentially a nanoscopic event. It was observed that because of an inability to adequately account for the microstructure at the required scale, the continuum models largely depended on a stochastic treatment of the subject.
Continuum models: mainly stochastic
Because the triggers for the initiation of metastable pits and their repassivation or transition to the stable pits were poorly understood deterministically, a stochastic model that was proposed by Williams et al. 36 based on a treatment by Shibata 180 has been frequently used. Experimental data analysis is required to fit this model to a particular system. 36 However, this model simplistically assumed that each micro pit had an equal chance of propagating into a macro pit, which disregarded the role played by the microstructure, the local microenvironment and any aspect of the pit itself, e.g., the narrowness of the mouth or the existence of a pit cover. Nevertheless, it is interesting that the likelihood of interactions between the metastable pits, which were recently observed using in situ techniques at the micrometre scale, 170,181 were actually predicted by a stochastic model by Wu et al., 37 which pre-dated the observations. By assuming through a ‘memory effect function’ that each pitting event will influence subsequent events and the influence would exponentially decay with time, that models could successfully reproduce the current transients that were recorded during the metastable pitting of an SS alloy and an aluminium alloy (however, it must be noted that the current transients may also be caused by events such as trenching or de-alloying). This temporal model was subsequently extended to a spatiotemporal version by Organ et al. 182 However, the model remained limited by the assumption that the surface was homogeneous without any preferred sites for nucleation because the microstructural effects were not modelled. A model developed by Walton et al. 183 (which is discussed later in the Stable pit initiation section) appears to be one of the first model that accounted for the active/passive transition based on the potentials. The CA models 184,185 that addressed metastable pits concluded that their growth is controlled by the anodic dissolution probability. Other models of note are by Malki and Baroux 186,187 and Hoerle. 188 In summary, the continuum models that accounted for metastable pitting were stochastic in nature and did not consider the influences of microstructure or other factors such as ions (the Cl− ions were neglected). They also did not account for the non-random nature of some features such as the intermetallic particles in some aluminium alloys, which were found in clusters instead of being arbitrarily distributed. 70
Proposed improvements
The present authors did not find any atomistic model that addressed metastable pitting. The common drawback among the existing continuum models is that none of these consider the microstructural effects despite their significant influence. 189 Therefore, an MSM should comprise a mesoscale model that adequately describes the microstructural features, which affect the metastable pitting, including the mechanism of active/passive transition if possible. An MSM should also be able to handle the differences in the behaviour of the current transients between systems, such as SS and aluminium (see Ref. 190), and such events as cooperative spreading. 169,170 In addition, because the pit nucleation events occur at the nanoscopic scale, 10 an MSM that incorporates metastable pitting should preferably include a model at this length scale, but a stochastic approach may be used for the initiation phenomena until such model is available.
Stable pit initiation
Pitting is caused by the localised failure of a passive oxide film 191 or the selective dissolution of a grain boundary or an inclusion 50 on a metal surface exposed to an electrolyte, and it is characterised by accelerated corrosion in the local region. For instance, it is generally agreed that anodic dissolution of MnS inclusions results in a change of the local solution composition near the inclusions, resulting in a condition where the passive film on the SS surface can no longer be sustained. 192 In general, most pits nucleate if the potential of the alloy surface is above the nucleation potential (or film breakdown potential or pitting potential) for the local electrochemical environment. After an incubation period, a pit propagates if the surface potential remains above the pitting potential. Especially in alloy systems involving SSs 99,167 and aluminium, 88 some metastable pits nucleate at potentials that are hundreds of mV below the OCP and eventually transition into stable pits (the Metastable pitting section). Therefore, the proposed MSM should allow either path for pit initiation. However, there are unanswered questions about the exact nature of the events that trigger pit initiation 79,99 and the mechanisms associated with film breakdown. 172 According to Kempf et al., 79 highly sophisticated techniques are required to study pit initiation events on sub-micrometre scales. High-resolution observations are, however, increasingly becoming commonplace, allowing informed theories to be developed. For instance, Marcus et al. 28 observed experimental data on the nanometre scale and proposed mechanisms for pit initiation at inter-granular regions. Other similar observations were reported recently by Magnussen et al. 163 and Williams et al. 99 Reviews of the critical factors affecting the pitting corrosion of pure metals and alloys are readily available, e.g., Refs. 115, 172 and 193.
The current review determined that in the absence of a consensus on how pits nucleate, various continuum models have taken different approaches. While the results of atomistic models are encouraging, additional experimentation on smaller scales is required to facilitate a convergence of views on this controversial topic.
Conventional theories: insufficient mechanistic detail
Two conventional theories encapsulate the traditional deterministic criteria: (a) critical solution chemistry theory (CSCT) or critical crevice solution theory (CCST) 194–196 and (b) Ohmic resistance-based IR drop theory (IRDT). 63,197 According to CSCT/CCST, passive film breakdown occurs when the pH and the Cl− concentration reach a critical state. However, according to IRDT, localised corrosion starts abruptly when the electric potential drop (IR) between the mouth and the interior of a pit or crevice is large enough to activate anodic potentials. Shortcomings of IRDT have been discussed by Frankel and Sridhar 115 and others (e.g., Refs. 198 and 199). Although CSCT/CCST has been supported overwhelmingly in the past, a recent attempt 63 has been made to develop and validate a model that unifies CSCT/CCST and IRDT. However, all of these theories are for pure metals and ignore the seminal role played by microstructural inhomogeneities (including surface roughness) on pitting initiation on engineering alloys. For example, neither of these theories include the preferential dissolution of select alloy phases as a cause of pit initiation.
Continuum models: multiple approaches
As mechanisms relevant to pit initiation take place on the atomic scale, the continuum treatment involves significant approximations and assumptions. In addition, the absence of a consensus view on pit initiation is reflected in the variety of approaches that have been used to model pit nucleation. Many of these studies have been reviewed by Sharland, 62 Frankel, 115,172 Kennell et al., 63 Papavinasam 200 and Anderko, 15 but a small sample of this research is discussed here. Anderko 15 reviewed several theories for passivity breakdown and concluded that all of them share a common theoretical result: the passivity breakdown potential varies with the logarithm of the concentration of aggressive ions, which is confirmed by experimental data. He also notes that, while this observation is accepted widely, its generalisation to systems with multiple types of aggressive and inhibitive ions is not obvious. Given the poorly understood nature of pit initiation mechanisms, it was convenient to use a stochastic approach to model pit nucleation (e.g., Refs. 38 and 34–37). Hybrid models (discussed in the Single scale models section) typically take a stochastic approach to model pit initiation (e.g., Refs. 46–48). These formulations calculate quantities such as pit birth rates based on probabilities or assumed statistical distributions rather than physically viable mechanisms. A common feature of stochastic models is that physical mechanisms (e.g., for passive film breakdown and pit initiation and growth) and microstructure are not included explicitly. These considerations limit the applicability of stochastic pit initiation models for alloy design. The pre-eminent deterministic models are the 1D steady-state models by Galvele 194–196 , the proponent of CSCT/CCST, which influenced deterministic thinking for decades by providing a step change in the ability to rationalise experimental results in various systems. However, as Newman 201 notes, the Galvele approach needs minor revisions to include highly concentrated metal salt solutions in pit nuclei. Other significant deterministic models are those of Alkire and Siitari, 202 Sharland, 203 Laycock and Newman, 204 Cong et al. 205 and Walton et al. 183 The transient 1D model by Walton et al. was one of the first that considered the electrode kinetics of both cathodic and anodic reactions with active/passive transitions. An electrode kinetic model developed and validated by Mccafferty 113 took into account the adsorption of chloride ions on aluminium oxide surfaces, the penetration of chloride ions through oxide films, and the localised dissolution of aluminium at the metal/oxide interface in consecutive one-electron transfer reactions. The 1D pseudo-steady state model of Webb et al. 192 modelled the influence of MnS inclusions on SS surfaces. However, all these continuum models suffer from the shortcomings discussed in the Conventional theories: insufficient mechanistic detail section.
Atomistic models: showing promise
Decades ago, Williams et al. 206 attempted to develop an atomistic model of pit initiation on random binary Fe–Cr alloys. They added a scheme for passivity breakdown based on CCST to existing atomistic models 207,208 by postulating that passivity breakdown corresponds to the critical chemistry necessary for the activation of the alloy. Their model identified the most important factors for pit initiation on SS: (a) the dissolution probability of Cr atoms; (b) the alloy composition, which determines the cluster size distribution; and (c) diffusion and migration within the cluster volume. A more recent atomistic work by Bouzoubaa et al. 18 has proposed tenable mechanisms of passivity breakdown. Currently, experimental observations are being made on the atomistic scale: e.g., the recent observations of aluminium oxide by Zavadil et al. 105 Rashkeev et al. 209 have performed first-principles quantum-mechanical calculations to provide an atomistic understanding of corrosion initiation on Al under atmospheric conditions. Their results suggest that atomic hydrogen penetrates oxide films and causes structural damage in oxides and at Al/Al2O3 interfaces. To summarise, atomistic modelling increases the understanding of pit nucleation by modelling mechanisms on their characteristic scales. Therefore, atomistic models are most suitable for modelling pit initiation.
Proposed improvements
Nucleation events are best modelled on atomic scales, and appropriate models have started to become available. These models, however, are still in their infancy and only explore probable mechanisms for pit nucleation, which remains a controversial topic. More investigations, like the XPS studies and MD simulations of amorphous Al2O3 by Chang et al., 210 may illuminate the relationship between oxide structures and passivity breakdown. First principle quantum-mechanical calculations such as those carried out by Rashkeev et al. 209 and Scully et al. 171 may provide an alternative to experimentation until better techniques become available for observing pit initiation events. However, until a deterministic model is developed for pit initiation, MSMs on localised corrosion may use stochastic models. It is necessary to prescribe the microstructure to apply the early deterministic models to the selective dissolution of inclusions/second-phase particles at rates based either on empirical polarisation curves (e.g., Ref. 50) or dissolution models (e.g., Refs. 25, 49 and 211). The effect of microstructural roughness can be incorporated by drawing from, for instance, the empirical relationship between surface roughness and pitting potential (e.g., Ref. 212). If continuum deterministic approaches are adopted until models on mesoscopic scales are available, other approximations may be required, for example, Olson et al. 213 They considered that surface energy must affect pit formation and treat pitting corrosion as a nucleation and growth process where surface energy promotes an activation barrier.
Pit or crevice propagation and rate
Pit growth is autocatalytic in nature in that alterations in local conditions 57 promote further growth of a pit in its propagation stage. (These environmental changes are considered in the Changes in electrolyte chemistry in pits section.) The spatial separation between the anodic and the cathodic reactions results in a negative pH gradient between the pit and the film that sustains the electrochemical reactions. Factors that control pit growth rate are discussed by Frankel. 172 While the initiation and early metastable growth of a pit are activation controlled (Butler–Volmer or Tafel kinetics equations), pit or crevice propagation above the pitting potential is mass transfer controlled 199 (Nernst–Planck equation). For an activation-controlled reaction, the current density is plotted on a polarisation plot (potential E v. log (current I)) or Evans diagram. Current density in a pit is a measure of the corrosion rate in the pit and, thus, a measure of the pit penetration rate. 57 Pioneering modelling work based on CSCT/CCST by Galvele 194,195 showed that a critical value for the product of the current density i corr with the pit depth x, the pit stability product = x.i corr, may be found in terms of the concentration of the ionic species at the bottom of a one dimensional pit. This critical x.i corr value corresponds to a critical pit acidification condition for sustained pit growth, and it can be used to determine the i corr value necessary to initiate or sustain pitting at a defect of a given size. 57 However, because Galvele's treatment explicitly ignores the effects of cathodic reactions inside the pit, it is only relevant at potentials well above the OCP of the metal in its critical localised solution. A topic that remains controversial is the degree of influence exerted on pit propagation by salt films that form on electrodes when the dissolving cations and, say, Cl− ions combine at saturation concentrations of the salt in solution. 204 Another unresolved subject is the formation of lacy covers on pits on SS that limit diffusion at pit mouths and thus help to maintain the critical chemistry in the pits. Some authors have proposed that this phenomenon is caused by the strong dependence of dissolution kinetics on local dissolved metal ion concentration (e.g., Ref. 198).
The present review found that only continuum models were available for propagation events. Although these models are able to model pit propagation reasonably well (as it is a relatively higher-scale event), they can be applied only indirectly using empirical parameters that account for associated mechanisms rather than explaining the direct influence of causal factors such as microstructure. Thus, continuum models are not sufficiently general to be incorporated into a truly generic MSM. Additionally, a deep understanding of electrode kinetics that would facilitate the development of reliable atomistic models was lacking.
Continuum models: the only option available
Because the propagation mechanism is better understood than the nucleation mechanism, more continuum models are available for pit or crevice propagation. Most models, including hybrid versions, follow the deterministic path for propagation. However, a number of stochastic models also exist. Sharland, 62 Turnbull, 214 Frankel, 172 Scheiner and Hellmich, 199 Kennell et al., 63 Papavinasam 200 and Macdonald and Engelhardt 215 have reviewed traditional models and formulations for propagation-related mechanisms, and only a selection of these models are reviewed here from a multiscale modelling perspective. Stochastic models typically calculate expected values of quantities such as the induction time or the number of stable pits as functions of defined parameters such as the nucleation frequency, the survival probability and the critical age. Then, through the interpretation of measured current transients, values for the parameters are retrieved. Stochastic models of Williams et al., 36 Valor et al. 35 and Alamilla and Sosa 33 are noteworthy examples. While none of these models explicitly consider the microstructure, the previously mentioned stochastic model for inter-granular corrosion developed by Zhang et al. 68 (the Continuum models: rudimentary description of microstructures section) utilised a brick wall model with a rectangular 2D geometry for aluminium grains. This rather simplistic description appears to be the closest stochastic approach published for modelling alloy microstructure. To summarise, stochastic models have not explicitly accounted for critical factors. Rather, stochastic models have relied on empirical parameters that were used subsequently to estimate various quantities associated with propagation. Deterministic models invariably followed either the CSCT/CCST or the RDT approaches. Often, early models (e.g., Refs. 160, 194–196, 202, 216, and 217) were developed for idealised 1D geometries, supported only the steady state, and contained several simplifying assumptions. Sharland and co-workers 30,203,218 and Engelhardt et al. 219 developed some noteworthy models, but Walton et al. 183 appear to be the first to develop and validate a transient model applicable to a wide variety of metals and electrolytes and supported by different kinetic rate equations. Laycock and White 198 developed a 2D FEM model that successfully recreated the lacy covers observed on pits on SSs. The deterministic model of Turnbull et al. 48 contained statistically distributed parameters for the pit growth equation. When Laycock and White applied the previously developed propagation models 198 in their hybrid version, 47 they accounted for different SS alloy compositions by changing the OCP for each alloy and for surface roughness by altering the distance of the initial spherical pit beneath the metal surface in the Tafel equation for anodic dissolution. Additionally, the deterministic propagation equation in the hybrid model of Engelhardt and Macdonald 46 for manganese steel in CO2-acidified seawater and Al in tap water contains empirically fitted parameters rather than values obtained from physical mechanisms. Such empirical modelling has been carried out for Al alloy AA7075-T651 using neural networks by Cavanaugh et al. 41 Investigating two orientations of metal exposed to varying temperature, pH and [Cl−], those workers concluded that pit growth generally followed t 1/3 kinetics. To summarise, most models used the 1D approach to predict pit size and transfer processes, 215 and existing continuum models of pit propagation have not explicitly considered the effects of causal parameters such as alloy microstructure, oxide films or surface roughness on propagation rates. Rather, these parameters have been incorporated using quantities such as the OCP or the exchange current density (in the Butler–Volmer equation, for instance) that were obtained empirically. The lack of generality makes it difficult to use such models in an MSM for designing alloys or corrosion-resistant systems.
Proposed improvements
There are very few atomistic models of pit propagation in the literature. The probable reasons for this situation are (a) the continuum models have tackled this phenomenon reasonably well on the continuum scale and/or (b) the computational cost does not justify applying atomistic simulations to phenomena on mesoscopic scales or continuum expanses. However, it is likely that atomistic methods will be used increasingly to simulate propagation and extend the boundaries of knowledge. There is a need for advancement in the fundamental theoretical understanding of electrode kinetics before reliable atomistic models can be developed. For instance, Macdonald and Engelhardt 215 recently noted that while Tafel constants may be calculated ab initio, the exchange current density is almost always measured experimentally because theory is not sufficiently developed to calculate this quantity from first principles. This is because Tafel constants contain relatively little kinetic information and the transfer coefficient is usually assumed to be 0⋅5 corresponding to a presumed symmetric barrier, whereas the exchange current density is derived from a highly kinetic and inherently complex set of mechanisms involving solvent reorganisation, which are still poorly understood. 220–222 Once an understanding of the charge transfer reactions at the electrode/electrolyte interface and related mechanisms is established on the atomic scale, however, rate equations such as Butler–Volmer or Tafel with empirically determined parameters to describe boundary conditions at the continuum scale will unnecessary. Recent phenomenological models 146 analyse charge transfer in solution in terms of a number of components and effectively refine the electrochemical scale. Lower-scale models will manage the difficulties with mesh refinement on finer scales to better describe, for instance, small-scale material removal or large concentration gradients near the electrode/electrolyte interface under diffusion control. In a nutshell, pit propagation can be modelled reasonably well on mesoscopic scales in the absence of a lower-scale model. In this case, the incorporation of microstructural details at the electrode/electrolyte interface should be considered in the medium term. However, because the atomistic description gives the best resolution, efforts should be made to incorporate atomistic-scale models in advanced versions of the MSM. Development of such models may remain a challenge until issues with computational loads are resolved through hardware and/or software improvements. Finally, the potential use of mesh-free methods for pit propagation should also be explored because these methods are well suited for modelling moving discontinuities.
Changes in electrolyte chemistry in pits
The concentrations of species in a pit are affected by electrochemical reactions such as anodic dissolution and cathodic reduction at the electrode/electrolyte boundary, chemical reactions such as precipitation (see the Precipitation of corrosion products section) and dissolution of O2 and CO2 at the electrolyte/air boundary as well as dissociation and hydrolysis in the aqueous environment. As a pit grows and the pit depth exceeds the width of the pit mouth, diffusion is restricted between the confined localised pit volume and the bulk solution. This results in the depletion of consumed species (e.g., dissolved oxygen) in the pit but the enrichment of dissolved metal ions. Such enrichment results in the migration of anionic species such as Cl− from the bulk to preserve electro-neutrality and the hydrolysis of the dissolved metal ions that releases H+ ions in the pit and lowers the pH. This acidic chloride environment is aggressive to most metals and tends to prevent repassivation and promote pit propagation. Further information about this subject may be found elsewhere. 57,61,223–226
The current review established that the only models on the continuum scale were available, and the lack of resolution as well as simplifying assumptions limited the application of these models.
Continuum models: purely deterministic
Stochastic modelling of pitting is not commonplace. Sharland 62 reviewed some of the early studies and concluded that no single model predicted (qualitatively or quantitatively) all the available experimental observations. The Walton model 183 predicted concentration profiles more accurately than previous versions. Evitts 227 carried out an extensive review of both steady-state and transient models that dealt with changes in chemistry, including the studies by Sharland 30,203,218 and Walton, 183 and concluded that most models assumed isothermal conditions and constant bulk solution chemistry. Macdonald and Engelhardt 215 noted that most models are 1D and erroneously neglect the potential drop outside the corrosion cavity, leading to significant errors in the prediction of damage. The White et al. 228 model relied on the Nernst–Planck equation rather than the electro-neutrality condition for the calculation of potential differences and predicted concentration profiles for an SS system. Laycock and White 198 calculated the local chemistry in an SS pit and accounted for several relevant criteria including a moving boundary due to propagation. More recently, Kennell et al. 63 developed a combined CCST/CSCT-IRDT model that provided the best agreement yet with the experiments of Alavi and Cottis, 229 surpassing the models of Sharland, 218 Walton, 183 White 228 and Evitts, 227 and introduced the possibility that such a combined approach may be superior to the CCST/CSCT-only path. Taxen and Persson 230 considered concentration changes due to the evaporation of aqueous electrolyte and used a moving mesh, but the application of the dilute solution theory was discontinued beyond a certain loss of volume. Finally, Heppner et al., 231 noting that most models assumed a dilute solution, carried out theoretical calculations for non-ideal solutions where ionic interactions could no longer be neglected. Their work, which used the Pitzer model, 232 simulated crevice corrosion evolution including species concentration changes and delivered better predictions than those models that did not account for ionic interactions. Thus, the use of the Pitzer model in the MSM should be explored, especially for non-ideal solutions. Moreover, in actual solutions, the activities of species can change not only with temperature but also with pH values. 15 Even for dilute solutions, traditional models relied on estimating activity coefficients from theoretical models (see Refs. 15, 183, and 233), and advanced modelling on this front should be considered. Another common feature in traditional modelling was the lack of data on chemical reaction kinetics. Therefore, several assumptions were made. For instance, Sharland and Tasker 218 assumed that the chemical reactions occurred at very high rates compared with other phenomena such as diffusion. While many such assumptions may be justified for continuum models, they are invalid for atomic scale models. To conclude, the existing models that describe chemical changes in a pit all belong to the continuum scale and thus lack detail on the mesoscopic scale for some relevant events discussed below.
Atomistic models: may be required for non-dilute solutions
Lower scale atomistic models that describe changes in solution chemistry are rare in the corrosion domain. This may be because the chemical changes are largely a response to other events rather than being causal events themselves. This means that, if events at electrolyte–electrode and electrolyte–air boundaries were modelled with sufficient accuracy and the important chemical reactions were properly accounted for, the chemical changes in the pit would be adequately modelled at the continuum scale without the need for a computationally intensive atomistic treatment. Nevertheless, as the theoretical work of Heppner et al. 231 showed, ionic interactions may become significant in non-dilute solutions where dilute solution theory would break down. Such cases might benefit from atomistic-level computations, which can provide the necessary resolution to model the interactions in more detail. For instance, in the multiscale framework, an atomistic-scale model can be used to calculate the empirical parameters (e.g., the activity coefficients and the osmotic coefficient) for the Pitzer model 232 for deployment at a higher scale, although these parameters have been experimentally derived for some systems. 234 Furthermore, the following events may be better modelled at a scale lower than the continuum: (a) the influences of alloy microstructure on local chemistry (e.g., through space-dependent dissolution) or vice versa (e.g., the local chemistry can preferentially attack metallurgical defects); (b) the passive film or its influence on the chemistry; (c) the microenvironment defined by the microscopic surface roughness; or (d) interactions between pit sites. Such detail is desirable in an MSM that will be used in a predictive role in alloy design. Molecular dynamics simulations of non-ideal solutions have been performed, 235,236 and perhaps similar studies could be used to explore the concentrated solutions that develop in pits.
Proposed improvements
In non-dilute solutions, phenomena such as ionic interactions within the electrolyte may not be neglected and ideally are modelled on the atomic scale to arrive at an optimum generic approach that could be used widely. Another area that needs to be strengthened is the quantification of reaction rate constants for some reactions. Although these constants could be obtained experimentally for some reactions, other extremely rapid reactions present difficulties in the determination of rates because reliable experimental measurements cannot be made easily using traditional methods. 237 In the absence of dependable measurements, the application of theories such as 238 the collision theory or transition state theory may be attempted for estimating reaction rates, and subsequently rate constants (see Ref. 239). Alternatively, these may be obtained from the application of first principles (e.g., Ref. 19). Similarly, it appears that no current document has a comprehensive listing of all relevant parameters such as those above and others such as dissociation constants and solubility products that a modeller could conveniently access, although some handbooks contain samples of these. Therefore, there are opportunities for developing atomistic models capable of estimating these quantities.
Precipitation of corrosion products
During localised corrosion, some salts precipitate on the corroding substrate as a result of reactions between dissolved anions and species in the electrolyte in the pit or crevice (see, e.g., Ref. 85). Precipitation occurs when the product of the ionic reactants exceeds the solubility product. The corrosive microenvironment under surface deposits is very different from the bulk solution. In particular, the pH of these microenvironments tends to be very acidic. The formation of acidified microenvironments is related to the hydrolysis of corrosion products and the formation of differential aeration cells between the bulk environment and the region under the deposits. 59 Precipitation of salts on electrode surfaces can affect the anodic and cathodic reaction rates by modifying the extent of the areas that are in contact with the electrolyte and/or increasing the resistance of the passive film to charge transfer. For example, the dependence of the Zn corrosion rate on pH is determined by the corrosion products. 61 Conditions controlling the thickness of products in zinc 61 and SS 240 systems are discussed elsewhere. Given the rate-controlling nature of the precipitates and the alterations to solution chemistry, an MSM should have the facility to adequately account for corrosion products.
The present review found that only continuum scale models were available to address precipitation, and these models contained sweeping assumptions such as homogeneous electrodes to describe what are, in essence, atomistic phenomena that are influenced by the heterogeneous electrochemical behaviour of electrode surfaces.
Continuum models: severely lacking in detail, but the only type available
As with the changes in pit chemistry (the Changes in electrolyte chemistry in pits section), only deterministic models are relevant for describing precipitation. Models that account for precipitation include those of Gravano and Galvele, 196 Sharland and Tasker, 218 Sharland, 203 Walton et al. 183 Laycock et al. 25 and Laycock and White. 198 These models provide different mathematical treatments of precipitation. For instance, Gravano and Galvele 196 treated precipitating solids as colloidal diffusing species in their work that focussed on solution chemistry immediately after film breakdown. The 1D transient model constructed by Sharland 203 was one of the first models to predict the evolution of solid phases (corrosion products) as a function of time and space in addition to solution chemistry and electric potential. All the above models assumed that the precipitation of the metal hydroxide occurred when its concentration reached the saturation level in the solution; Laycock and White, 198 however, allowed up to 10% super-saturation to occur owing to their additional assumption that a hydroxide salt film could precipitate only on an existing salt film or on the corroding metal. A slightly different approach was followed by Taxen and Persson, 230 who assumed that precipitation occurred when the activity coefficient for any solid species in solution exceeded unity. Another aspect of traditional modelling has been the limited use of data for rates of precipitation by many models (e.g., Ref. 218) based on the assumption that rates of precipitation are much higher than other events such as diffusion. It was necessary to make these assumptions in the traditional models due to the general lack of data on chemical kinetics and rates of precipitation, 218 which has remained the case to this day. 215 Some workers such as Farrow et al. 110 worked around this issue by using theory 241 to estimate the rates. Some works such as those by Nesic and Lee, 242 Sun and Nesic, 243 Anderko 15 and Taxen and Persson 230 dealt with the subject in the sphere of general, rather than localised, corrosion. Also worthy of mention is the work of Tidblad et al. 244 who used the well-known GILDES model 241 for atmospheric corrosion to predict the formation of corrosion products on copper substrates exposed to aqueous species. The GILDES model solves the mathematically formulated transformations and transitions that occur in the six relevant ‘regimes’: the gas phase, the interface between a gas and a liquid, the liquid phase, the deposition layer, the electrode region near the surface and the solid phase. With the exception of studies such as that of Farrow et al. 110 and the GILDES model, 241 none of these models have accounted for the well-known influence of surface pH on precipitation by simulating the dissolution of precipitate layers in certain pH ranges: so these models were limited to pH ranges where the products were stable. None of these models also took into account the effect of microstructure and were unable to provide the details required to model mixed corrosion products, such as different oxides of alloy components or their spatial distribution that depends on microstructural features besides alloy composition. Additionally, none of these models explicitly accounted for the passive films. Because the electrode was assumed to be homogeneous on the macroscopic scale, the available models do not contain the details that a prospective microscopic or lower-scale model would be capable of furnishing. Thus, the available models need fine-tuning from the perspective of incorporating precipitation algorithms into an MSM.
Proposed improvements
The present authors did not find any atomistic-scale models that dealt with corrosion products, although, like oxides films (the Passivating oxide film, its thickness and its porous nature section), the usually ultrathin and porous nature of precipitate layers make them classic examples of why a multiscale approach should be taken for modelling localised corrosion. The atomistic scale provides the ideal framework for modelling transport at not only the interfaces between the products and other components such as the passive film or the metal itself but also through the products themselves. This is also the most suitable method for modelling interactions between the electrolyte and the products including adsorption. In addition, bonding at interfaces may be modelled elegantly using an atomistic approach. Thus, there is scope for building models of corrosion products at the lowest scales in the medium to long term. These models should take into account the microstructural inhomogeneity of the metal substrate and the oxide when predicting the space-dependent structure and thickness of the precipitate layers. It may be possible to develop atomistic precipitation models for corrosion based on precipitation kinetics-related models that are becoming available in other fields, e.g., solidification-atomistic 78 and hybrid atomistic-kMC. 245 Additionally, the inverse of precipitation (dissolution) of salt films has been studied recently using MD, 246 and this approach may be examined for developing a basis for precipitation models of corrosion.
Repassivation
Under certain circumstances, the increasing corrosion partial current that follows a nucleation event may cease. This is attributable to repassivation and is due to the loss of the active surface on the electrode. Despite the autocatalytic nature of pitting, even large pits can stop growing or die. 57 While the repassivation of the corroding surface stifles the growth of pits, the increasing Ohmic potential drop along the depth of the pit also results in the pit being stable at a low anodic current density at the bottom. This is because, as the pit deepens, the diffusion of the cations out of the pit reduces, thus decreasing the rate at which these ions dissolve from the substrate. At some point, the pit stops growing and is considered ‘dead’. The repassivation mechanism/s for metastable pits are not known for certain. 247 Relatively recent high-resolution experimental works, however, are beginning to illuminate some of the mechanisms involved in repassivation. For instance, Chidambaram et al. 248 have observed, through synchrotron infrared microspectroscopy and secondary ion mass spectroscopy, that the slow migration of ions from the surface of AA2024-T3 alloy protected by chromate conversion coatings to the scratch-exposed metal surfaces leads to the repassivation of the metal. A similar mechanism might be operating in metals protected by oxide films. Furthermore, the quantitative effects of the alloying elements on kinetics have been studied by some workers. For example, Cho et al. 249 have quantified the influence of alloying elements on repassivation kinetics. Data on repassivation kinetics remains scarce. However, repassivation kinetics are considered to be a critical factor in determining the resistance of metals and alloys to localised corrosion 250 and, consequently, in influencing the accumulated damage. 104 Thus, there is little doubt that any MSM built for use in the design of corrosion-resistant alloys should be able to simulate phenomena relating to repassivation.
This review found mostly continuum scale models that relied on Pourbaix diagrams for equilibrium conditions and mostly empirical data for non-equilibrium situations, and thus had little general applicability. This review also highlighted the lack of atomistic models and experimental data on the smaller scales.
Continuum models: inadequate
Traditionally, conditions under which repassivation occurs have been determined by the use of Pourbaix diagrams. 247 However, this phenomenon has been addressed by a handful of empirical and continuum mathematical models as engineering situations deviate significantly from the equilibrium-based scenarios. Examples of empirical models include that of Song et al. 251 However, as the empirical models do not explicitly incorporate the influence of, for instance, the environment or material properties, they cannot be utilised as generic tools. Stochastic modelling works have attempted to incorporate the repassivation phenomenon using probabilities. For example, in the model by Williams 36 an assumption regarding the probability that a metastable pit will repassivate was made. However, these stochastic models, like their empirical counterparts, do not explicitly account for mechanisms leading to repassivation. Among deterministic models, the Walton et al. model 183 appears to be the first that is capable of simulating repassivation, as it supported the active/passive transition. Laycock et al. 25 assumed that repassivation would occur if the diffusion of metal ions away from the electrode exceeds their production through dissolution, in which case the ion concentration tends to zero. Anderko et al. 252,253 developed mechanistic models based on statistical thermodynamics. Their models calculated the repassivation potential for selected SS and Ni-base alloys whilst accounting for the influence of solution chemistry (aggressive species, oxyanions and inhibitors), temperature, competitive dissolution, adsorption and oxide formation. In particular, the models predicted the transition from the concentration range where localised corrosion was favoured to the region where inhibition was expected. Some of the parameters in the Anderko models 252,253 were experimentally obtained. A PDM 26,104,215 was capable of explaining the experimental observations of Ahn et al. 254 for the repassivation kinetics of Ti. However, as noted by Wu and Celis, 255 HFM is the most widely used explanation of repassivation kinetics, although its validity has been questioned by some. While some experimental works (e.g., Refs. 255–257) have supported the application of HFM, Wu and Celis 255 caution that HFM may not be physically realistic at very short times following depassivation. To summarise, while continuum scale models exist, they are inadequate for modelling repassivation because it is known that lower scale factors influence this phenomenon. For instance, second-phase particles or inclusions in an engineering alloy would exhibit electrochemical potentials different from that of the matrix, and can preferentially passivate (or dissolve). Such resolution at the microscopic level is mandatory for an MSM aimed at alloy design.
Atomistic models and experimental data at this scale are scarce
Using MC simulations, Malki and Baroux 185 determined that varying the repassivation probability on the atomic scale on the pit walls can control pit growth kinetics (t n law), but the authors admitted that there was no experimental evidence to suggest that this unexpected prediction was tenable. This model appeared to be the only one in the open literature that dealt with repassivation, and the lack of models at this scale is probably explained by the scarcity of experimental data on this scale. However, atomistic simulations have been performed for the oxidation of metal surfaces, which is a critical step in repassivation, e.g., DFT work by Schröder. 258 Perhaps strategies found in these works can be applied to develop a model for repassivation.
Proposed improvements
More experimental observations on the atomic scale are required to understand the mechanisms that lead to repassivation in situations that do not involve the preferential dissolution of phases or inclusions. It therefore stands to reason that atomistic-scale models will be the most appropriate means to describe such phenomena. However, the existing models that account for repassivation have all been developed on the macroscopic scale. Additionally, no existing model has taken into account the role of the microstructure, for instance in the apparent repassivation of pits formed by selective dissolution. It is also necessary to determine through experimentation whether the presence of different phases or other microstructural features in the local neighbourhood would have any influence on whether a pit repassivates. Such influences may be modelled at the microscopic level and will enrich an MSM designed for alloy design.
Interactions with immediate neighbourhood
A passive surface exposed to an aggressive corrosive environment is usually riddled with numerous pits. Due to a competitive interaction process between these pits, only the strongest survive to grow. 104,259 A stronger pit, which generates a higher current, competes for a larger share of the charged species by attempting to expand its ‘living space’, the volume of electrolyte immediately adjacent to the pit. The stronger pit thus suppresses its weaker neighbours. In some cases, possibly when the competing pits are similar in strength, they coalesce. Recent high-resolution experimental observations 169,170 have suggested that an autocatalytic explosion of metastable pits may be possible, at least in SSs. Therefore, it is useful to reproduce these interactions in detail in an MSM so that corrosion sites are predicted realistically.
Despite the dominant influence of the electrochemical properties of the microenvironment surrounding a pit on determining the favoured interactions (which calls for lower-scale modelling), only continuum models were found during the present review. With greater understanding of pitting at the lower scales, it should be possible to model these interactions at the atomic level.
Continuum models: hybrid treatment
Only a handful of existing models address this topic, and this is probably due to (a) the lack of experimental data and associated understanding, which is only just beginning to filter into the open literature, and/or (b) the probable perception that such interactions may only be of secondary importance. Available models generally take a hybrid approach treating nucleation and interaction events stochastically but employing deterministic equations for the influence of the environment. One of the first models that accounted for interactions was the previously mentioned stochastic model of Wu et al., 37 in which a memory parameter, M, was incorporated to retain the influence of a given event over time. Lunt et al. 260 extended this model and their simulation results, which agreed with experimental data obtained by the same workers on an array of SS electrodes, showed that surface damage gave the highest M value. Organ et al. 182 extended the Lunt et al. model to 2D, and this model suggested that interactions among metastable pitting events can lead to the formation of clusters of pits. White et al. 261 developed an FEM model incorporating interactions as an extension of a previously discussed model by Laycock et al. 47 Their model confirmed that an existing pit would increase the probability of pits nucleating at nearby sites. Harlow and Wei 262 developed a stochastic model incorporating interactions for pit growth in Al alloys. Although not validated with experiments, the authors mentioned that its predictions were in qualitative agreement with observations. Significantly, the authors advocated a deterministic approach for a more realistic analysis of interactions. A mechanism of competitive interaction between pits at the early stage of their development was considered by Popov, 259 who assumed that the interaction occurred due to the hydration of metal ions by the solvent. Popov derived analytical equations and proposed a 1D model of interaction between two linearly connected pits. However, this model was not validated with experimental data.
Proposed improvements
Only a limited number of models have addressed the phenomenon of interactions between pits. All of these models were on the continuum scale, and the electrode surface was modelled as homogeneous, without preferred nucleation sites. Therefore, a stochastic approach was followed for nucleation events, and interactions were modelled by deterministic equations that incorporated the influence of the environment on interactions. Clearly, the assumption of homogeneity reduces the detail required in a model for alloy design. Therefore, a model at the mesoscopic scale should be developed for describing the interactions on their characteristic scales. It would be an advantage to have an atomic scale model to describe nucleation events. However, limitations of atomistic models addressed earlier in the Proposed improvements section with respect to non-equilibrium events must be adequately dealt with before dynamic processes such as interactions can be realistically modelled at that scale. In addition, although recent in situ experiments and modelling indicate that an existing pit would increase the likelihood of pits nucleating at adjacent sites, at least on SS, it would be desirable to have additional observations of different corrosion systems. Lastly, when more experimental data becomes available, replacement of the stochastic treatment by deterministic mechanisms would help interrogate the models and thereby increase our understanding of the controlling factors.
Summary
Figure 5 provides a summary of the approaches discussed in some detail in the preceding sections. Continuum approaches are shaded in red and atomistic in green.
Numerical strategies
In this section, issues unique to the solution of equations on multiple length and time scales are addressed.
Field variables at different scales
The primary challenges for modelling corrosion where different scales of time and length are linked can be classified broadly as formulational and computational. Formulational challenges arise when two or more apparently dissimilar modelling philosophies operating at different scales are combined to explain a phenomenon such as corrosion. It is a formidable task to combine the mathematical structure of one modelling approach with another while ensuring a seamless information exchange between the different models. Computational challenges, on the other hand, include appropriate hardware, efficient multithreaded parallel programming, approximation and fault tolerance, data visualisation and data transfer between various computing units. In this section, we concern ourselves primarily with formulational challenges of multiscale modelling and some computational challenges inherent in the development of an MSM for corrosion.
The central difficulty in coupling two methods on different scales is reconciling the definitions of computational variables. Two quantities are of primary interest: the total energy of the system and the temperature. The standard approach is to construct a potential energy functional and use its derivatives to obtain the forces necessary to evolve the system. However, Curtin and Miller 263 note that the fundamental problem in bridging the continuum and atomistic methods stems from the fact that the expressions for the total energy of the system are fundamentally different in the two methods. They observe that apart from the semi-empirical nature of the interatomic potentials employed in MD, which introduce uncertainty and approximations, the quantum energy, unlike its classical counterpart, cannot be partitioned into energies on a per-atom basis. This was reiterated by Makov et al. 264 who observed that, in the case of an MSM, two potential Hamiltonian formulations could be used for the atomistic and the continuum models. When more than one model Hamiltonian is used, it is normally assumed that the energies in the system are additive. However, the additivity of the energy is scale-dependent, being valid at large scales (continuum) and invalid at short scales (atomistic) due to the existence of long-range interactions and quantum non-locality. Thus, the width of the boundary zone where the models ‘shake hands’ must be chosen such that no forces are introduced artificially. Makov et al. 264 have described a ‘Learn on the Fly’ (LOTF) multiscale approach in which the entire system is represented in the high level, and the region of the low-level simulation is determined dynamically (i.e., continuously updated) by flagging the ‘quantum’ atoms according to a mixture of topological (bonding lengths and angles) and geometric (e.g., distance from the crack tip) conditions. The potential used to derive the forces in the boundary zone is fitted to the results of the low-level atomistic simulation, and this unique potential describes and conserves momentum.
On the other hand, Ganzenmüller et al. 265 observe that both the heat flux and the temperature are interpreted differently in continuum and atomistic domains. The continuum approach is based on discretising a continuous field of state variables at discrete spatial locations. The momentum, mass (and species), charge and heat fluxes are then evaluated between these nodal points. Many techniques including finite difference, 266 FE, 267 control-volume 268 and their hybrid variants 269 employ this scheme. Techniques such as smoothed particle hydrodynamics (SPH) 56 based on Lagrangian methods, where the evolution equations are not constrained by the need to track the movement of the fluid particles, also evaluate the fluxes using averaging techniques similar to the earlier methods. Molecular properties such as viscosity (internal friction) are usually provided as transport parameters to the simulations. The conservation equations usually are developed for mass (species), energy, momentum and charge separately and solved by effectively capturing the non-linear coupling between the variables. However, in MD, the kinetic energy of the interacting atoms depends on the momentum exchange because no other degrees of freedom except those associated with the particle kinetic energy are present and, thus, the temperature depends solely on the momentum. Ganzenmüller et al. 265 christen the average temperature in MD simulations as the kinetic temperature because it stems purely from the kinetic consideration of a large number of interacting atoms. They distinguish this from the continuum temperature, which in continuum models is defined using the concept of internal energy and heat capacity. They further assert that the kinetic temperature in an atomistic simulation relates to the average particle momentum, whereas the continuum temperature (even if coupled to the momentum conservation equation) serves only as a state variable without any relation to the momentum of the corresponding continuum integration node. Ganzenmüller et al. 265 further observe that a correct continuum-MD coupling algorithm must describe the heat flux between the two domains such that the continuum variable internal energy is linked locally to the MD particle velocity. They also advocate dissipative particle dynamics at constant energy (DPDE) as an ideal choice for the mesoscale coupling. Albeit an isothermal method, DPDE, has provisions for assigning an ‘internal temperature’ to each particle that can be related directly to the internal energy and the heat capacity, and the local DPDE thermostat is used in the MD domain to achieve dynamic equilibrium between both temperature definitions.
Modelling electric potential
Another closely related issue is the ‘conjugate problem’ 269 in which two or more regions (or phases) are involved and there is a ‘jump discontinuity’ in the material properties and state variables. In corrosion, we encounter two or more phases where some quantities must be defined unambiguously to ensure continuity. Electric potential is the only macroscopic variable that is present in both the solid and the solution domains, although a discontinuity is prescribed (i.e., the potential of the metal and the potential of electrolyte have different values) at the solid–solution boundary to quantify the electrochemical surface reaction using Butler–Volmer kinetics. The reason for these discontinuities is simply the incapability of the continuum models to describe atomistic details. In the continuum methods such as the control-volume finite element method (CVFEM), the mesh is constructed such that any given integration element falls in one and only one region, which ensures there is no ambiguity in calculating the face diffusivities in a given element. While the conjugate problem is based on a genuine phase-differentiation, the atomistic-continuum boundary zone is only a numerical artefact. Thus, while it is important to ensure that modelling the conjugate problem considers accommodating the atomistic details of the interface, it is equally important to ensure that the spurious forces at the ‘numerically created boundary zone’ do not contribute to the atomistic-continuum handshaking as well.
Candidates for future MSM and mathematical coupling of scales
Tan, 12 Elliott 5 and Curtin and Miller 263 have reviewed a number of multiscale methods with particular emphasis on fracture. Tan discussed stress-corrosion cracking and pitting corrosion and observed that stress-corrosion cracking is the result of sufficiently strong mechanical forces that separate chemically bonded atoms and also presented a case where hydrogen interacted with dislocations. However, the combined interaction of mechanical stress (developed due to deformation or loss of atoms) and electrochemical reactions underlying corrosion was not discussed in detail. Similarly, Tan discussed pitting corrosion in the context of MC simulations performed by Reigada et al., 32 who assumed that the probability of ‘tunnelling’ (metal oxidation) depended linearly on the local halide concentration and exponentially on the applied potential. Bartosik et al. 270 considered that pit nucleation is a rare event at the atomic scale and that a MC simulation is still far too large for currently available computational resources. They also noted that the complicated spatial and temporal oscillatory behaviour exhibited by metals undergoing passivation in solutions poses a strong challenge to the development of a unified approach for covering various aspects of corrosion. Thus, at the mesoscopic scale, they have proposed a CA model that is characterised by seven species: metal M, reactive site R, passive site P, electrolyte E, anodic dissolution site A, and cathodic site B. They enforced local rules and noted the interdependence of the A and B sites and the underlying connectivity in the metal matrix that ensures that the electrons lost at an A site are compensated exactly and simultaneously at a B site. Cellular automaton methods on mesoscales typically require huge grids, and establishing a connected path in the metal matrix for three-dimensional simulations is very time-consuming. This method, although in an early stage of development, shows promise for future MSM applications because it is one of the very few methods that accounts for global charge conservation, which is a stringent requirement for corrosion. This approach however has not yet accounted for the following phenomena: (a) the diffusion and migration of species such as dissolved oxygen in the electrolyte, which determines local corrosion rates; (b) the effect of metallic microstructure including local phase compositions on multi-phase/polycrystalline metals and/or alloys; (c) distributions of defects in the microstructure; and (d) effects of porous semi-conducting oxides (precipitated from the solution or natively formed) that affect the permeability/percolation of the electrolyte and host ORR on their surfaces, 271,272 thus enhancing corrosion rates to more than expected levels for metals such as iron and zinc. The CA models can be refined to an atomistic level and the atomistic-mesoscopic scale coupling can be achieved by refining the cells to match the atomistic surface morphology. This refinement could place a high computational load on the current CA models especially for corrosion on a contiguous metal surface. The CA models could be combined with control-volume from CVFEM techniques, which will account for reactions and ionic movement in the solution phase. Additionally, SPH is another method that can include localised precipitation. 273
Elliot 5 observed that the atomistic-mesoscale coupling can be performed by ‘coarse-graining’ at the boundary zones and integrating out redundant degrees of freedom. This is achieved either by forcing atoms onto a lattice or by grouping them into larger particles. The lattice material point method (LMPM) 12 or SPH 56 could be used for this purpose. In both LMPM and SPH, the continuum material point is modelled as an aggregate of atoms (‘particles’ or ‘atomic aggregates’), and the entire continuum is modelled as a collection of material points (or particles) although, unlike SPH in LMPM, a background mesh is always used. In both methods, a Lagrangian description is used to discretise the material into a collection of atoms whose motions characterise the deformation of the material. In LMPM, once the atoms are combined into larger particles, a quadtree or octtree method could be systematically employed to aggregate the atoms in the transition zone for 2D and 3D models, respectively. The stresses are calculated from velocities that are computed using Verlet algorithms. 274 However, Hoover 275 warned that the analogy linking the SPH to atomistic MD also suggests that the SPH representation of a continuum might exhibit the same chaotic instabilities that are present in atomistic systems. Ganzenmüller et al. 265 proposed a coupling strategy wherein they employed SPH for the partial differential equations (PDEs) in the continuum to which a region with atomistic length scales and corresponding particle dynamics was coupled and this region was described by classical MD employing a DPDE thermostat. Interpolations could help damp some of the oscillations encountered at the boundary points, but LMPM, although computationally expensive, seamlessly integrates with the framework of MD.
There is another interesting issue, mentioned briefly in the Proposed improvements and the Proposed improvements sections, which requires discussion. It concerns with the difficulty of linking electron transfers during non-equilibrium situations such as the formation of anode and cathode at the atomistic scale, where calculations are typically carried out for equilibrium configurations. As previously indicated (the Proposed improvements section), fluctuation theorems 165 may provide a pathway for dealing with such events because they enable quantitative predictions on fluctuations in small systems that are monitored over short periods to be made; therefore the fluctuation theorems allow thermodynamic concepts to be extended to apply to finite systems. They describe the statistical fluctuations in time-averaged properties of many-particle systems such as fluids driven to non-equilibrium states and provide some of the few analytical expressions that describe non-equilibrium states. Incorporating the fluctuation theorems in the MD simulations (which are formulated using a Newtonian approach and not using DFT) could provide a method to predict the evolution of time-irreversible electrochemical systems of molecular dimensions. Once these modifications have been incorporated, one could expect an electronic/charge distribution (thus non-equilibrium) to result from the calculations and subsequently the atomistic-mesoscale coupling could be done by ‘coarse-graining’ at the boundary zone by integrating out redundant degrees of freedom using LMPM.
Another problem that requires consideration is solving equations on different scales. Consider that anodic half-cell reactions constituting localised corrosion start at microscopic anodic sites and eventually spread on macroscopic scales. Often, the microscopic anodic sites couple with much larger cathodic areas on the continuum scale. Similarly, there is a vast difference in characteristic time scales between (a) the almost instantaneous phenomena such as electromigration, chemical reactions and electrochemical reactions; and (b) the decidedly slow processes such as diffusion. The PDEs in an MSM that describe such incongruent phenomena occurring over different length and time scales that are several orders of magnitude apart present numerical solution issues that are known as stiff problems. A stiff equation is a differential equation for which certain numerical methods for solving the equation are numerically unstable unless the step size is extremely small. The main reason for this issue is that the equation includes some terms that can lead to rapid variation in the solution. While generic solution strategies have been contrived to solve such problems, a complex set of equations, as in the case of corrosion modelling, requires a customised approach to obtain a stable numerical solution in a reasonable time frame. Developing such a successful solution strategy is part of developing an MSM. Incidentally, Macdonald and Engelhardt 215 have discussed alternative approaches to solving the system of equations containing chemical reaction and transport terms with vastly different characteristic time scales and thereby rates also. The numerical method employed by White et al. 228 separated concentration changes due to chemical reactions, which can have infinitely large rates of change, from concentration changes caused by transport so that the PDEs may be solved more easily. Walton et al. 183 and Sharland 203 decoupled equations for precipitation reactions from mass transport and corrosion equations on the basis of vastly different characteristic time scales. They used the same procedure when modelling changes in chemistry inside the pit. Thus, in addition to overcoming the formulational challenges associated with the linking of spatial scales, an MSM developer will need to resolve issues related to the different temporal scales. In the latter, the coupled equations will need to be solved using techniques that account for: (a) properly reconciled definitions of the computing variables (e.g., kinetic and continuum temperature), (b) stiffness and (c) instabilities that may propagate from the lower scales to the higher.
Some suggestions for future MSM features
Initial versions of MSMs on localised corrosion will have incremental improvements over current techniques rather than satisfying all of the lofty goals outlined in the Formulation of crucial phenomena and the Numerical strategies sections and will address only some of the fundamental challenges outlined therein. An enormous amount of modelling and experimental effort will be needed to develop an ideal MSM as envisaged here, which will be generic enough to apply to different alloy systems.
Modellers will first need to decide whether their MSM will need to be a three-scale model (e.g., Ref. 10) or a two-scale version (e.g., Ref. 75). That decision will hinge on the degree of resolution sought at the various levels and will be dictated by project goals. As argued in the Formulation of crucial phenomena section, a realistic MSM for the development of corrosion-resistant alloys and inhibitors should be coupled to an MSM on alloy solidification. It appears that a parallel framework (Fig. 2 and Ref. 9) is the most suitable option for this coupling as shown in Fig. 6 for a hypothetical case.
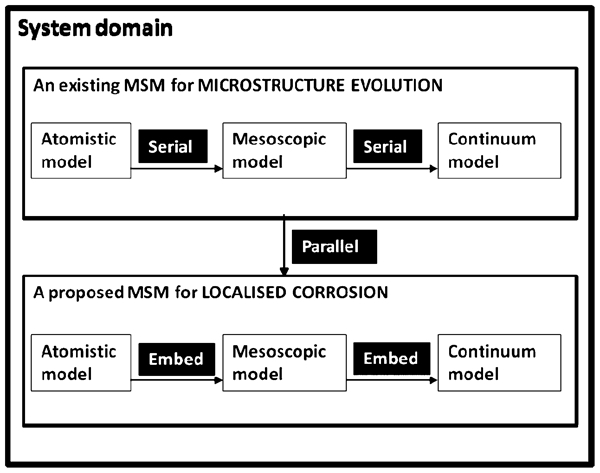
Different classes of integrating multiscale frameworks proposed for a hypothetical case where an existing multiscale model (MSM) for microstructure evolution is coupled with a proposed MSM for localised corrosion
As shown in Fig. 6, we favour an embedded framework (Fig. 2 and Ref. 9) for an MSM on localised corrosion. This approach will reduce the overall computational burden by limiting the volumes computed at the lower scales. A rather detailed conceptual MSM on localised corrosion envisioned by the present authors is presented in Fig. 7.
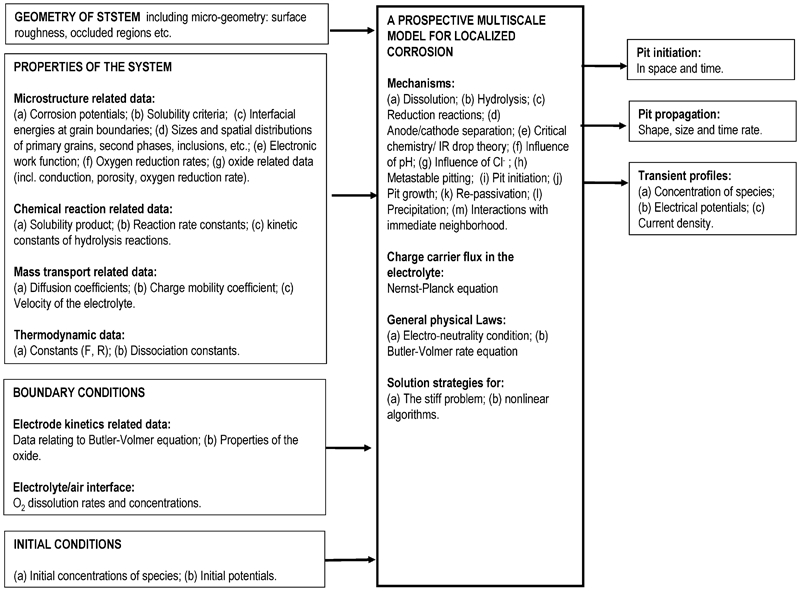
General description of a prospective multiscale model (MSM) on localised corrosion including potential inputs and outputs
In developing a barebones MSM for corrosion, the following processes must be envisaged:
metal oxidation
metal ion dissolution, diffusion, speciation and hydrolysis
oxygen reduction
OH-ion dissolution, diffusion and further solution reactions
precipitation of corrosion products from the solution
effect of initial oxide layer
solution chemistry (migration and diffusion of other ions like Cl− and inhibitors).
The favourable attack sites for corrosion on the metal surface are the defects and the grain boundaries. Hence, while developing an MSM for corrosion, it is imperative to model the surface atomistically using lattice techniques. However, unlike fracture, corrosion is basically a surface phenomenon that could potentially, in the long run, lead to a situation where the pits or crevices are large enough to cause material failure. Although apparently coupled, in the first version of the MSM we can delink the modelling of the electrochemical reactions from the modelling of material failure due to crack growth (which can be handled efficiently by many methods proposed in the literature 12 such as LMPM and the quasi continuum method). Instead, corrosion modelling could concentrate on the interface and the large surface area over which pits initiate, grow, interact, stay metastable and/or die. However, the surface of most active metals is covered by an initial oxide layer, and localised corrosion occurs when this oxide layer is damaged. The PDM, 26,276 in principle, addresses defects and has a subtle connection with MD methods in the solid phase, although the PDM treats defects in a macrohomogenous fashion. While it is tempting to argue that the PDM or its variant 276 can contribute to coupling algorithms, the connection between vacancy density and atomistic vacancies needs to be established through conservation principles. On the other hand, the interaction of an inhibitor with the metal surface is a vast subject that is currently gaining momentum. 126 While DFT methods have been employed primarily to understand how inhibitors interact with metal surfaces in solvated environments, the computational constraints dictate that this interaction, once well understood, must be only semi-empirically accounted for using MD methods that can later be coupled with one of the candidate continuum methods (FEM, CVFEM, SPH or LMPM) and mesoscale methods (CA and DPDE) described above.
Reactions
At an international workshop 13 attended by eminent electrochemists, it was concluded that progress in deterministic modelling will depend critically on progress made in the understanding of the reaction kinetics of crucial corrosion reactions, although specific reactions were not identified. The present authors discussed this in some detail in the Proposed improvements section. The workshop mentioned also identified (i) a chasm between fundamental studies that focus primarily on ideal surfaces; (ii) the challenges associated with real surfaces such as surface defects, surface films, adsorbed species and water molecules; and (iii) a lack of knowledge about the morphology and composition of porous films and the influence of alloying elements on film properties. Such knowledge is now beginning to appear in the literature thanks to state-of-the-art experimental techniques, e.g., in situ electrochemical scanning tunnelling microscopy, 103 which sheds light on the initiation of nano-scale pits on nickel surfaces.
Mass transport
It may be convenient to use the dilute solution theory 277 for the initial versions of the MSM, although this is not recommended for real solutions. Activity coefficients that account for the non-ideal behaviour of charged species at non-dilute concentrations should be used where possible.
Micro climate
As outlined in Fig. 1, the lower level and pitting models will need to be connected to models that can predict the ‘state’ of the exposed surface where local attack occurs. By state, we refer to the presence or absence of moisture, the chemistry of aqueous phases, and the temperature of the metal surface. The models derived by Cole et al. 278–281 that link surface state to climate parameters and sources and distribution of pollutants can be used or adapted for this purpose.
Conclusions
This review has highlighted both the complexity of developing MSMs and the possibility that a first generation MSM for localised corrosion that spans from molecular to component scales may be developed in the not-so-distant future. This MSM will have great utility for design, as it will allow the effects of atomic scale changes on the component performance to be determined. Thus, the molecular design can be optimised. Furthermore, processes that lead to the most rapid deterioration can be focussed upon at their characteristic scales for additional research or development.
Current design approaches are based on continuum models. We outlined how these models were based on assumptions of homogeneity and were consequently limited for applications to heterogeneous materials and processes that require a heterogeneous description. This was most evident in the cases of material heterogeneity, such as complex multi-phase microstructures, oxide films, paint films and corrosion products. Most present models assumed homogeneous properties for the material units. Where limited heterogeneity was permitted, it was introduced by linking two homogeneous units and ignoring the boundaries. An example is the application of the porous-electrode theory to model corrosion processes where the solid–fluid boundary was accounted for by superposing continua. On the contrary, molecular or atomic modelling in MSM will allow heterogeneities to be studied directly without the overbearing assumption of sweeping homogeneity, but at the same making necessary allowances for information exchange at the ‘handshake’ regions where the continuum and discrete models intersect. For example, percolative transport of fluid through the oxide pores and its subsequent interaction with the metal surface underneath is a strong candidate where percolation could be modelled using statistical considerations (i.e., partially disregarding the discreteness in the distribution), and the reaction of the fluid with the metal surface could be modelled by DFT or MD techniques. As in the case of material structure, atomistic and molecular modelling can examine specific interactions between atoms and molecules that can then be linked back into a continuum scheme.
There are, however, significant challenges to developing an MSM. These challenges include:
The overall formulation of the model so that processes and structures are modelled on appropriate scales;
The development of models at each scale that are computationally efficient and provide results of appropriate accuracy and resolution;
The linking of models on different scales in a computationally efficient manner, and
The validation of the model overall and at each level.
This paper described a structure in terms of overall formulation and presented a number of possible approaches to this problem. The main computational costs occur on the atomic or molecular scale. As outlined by Elliot, 5 efficient methods are required to accelerate molecular modelling and effective partitioning, and linking molecular and higher-level scales may be part of the solution. The exact method to link different scales probably will depend on the particular material and processes involved, and thus a tool-box rather than a prescriptive approach may be best. As discussed in the Bridging the nano-gap section, a ‘nano-gap’ still exists between molecular modelling and continuum modelling of inhibitor effects. Addressing this gap is essential because most of the observed electrochemical phenomena depend on the double-layer properties. We discussed some of these strategies for linking DFT and MD to expand the fine scale or developing phenomenological models of electrochemical processes supplemented by refined measurements to model finer scales. Lastly, the validation of the MSM will be critical at each scale and at the overall model level. We emphasise that more refined experimental techniques will be required for this purpose.
Footnotes
Acknowledgements
The authors thank Prof. Rudy G. Buchheit (Fontana Corrosion Center, Ohio State University) who generously contributed to several discussions on topics of interest during his visit that was supported by a Fellowship from CSIRO and provided feedback on this manuscript. In addition, the authors would like to thank Dr Michael Breedon (CSIRO) for discussions on molecular modelling and DFT. Highly useful suggestions were provided by Drs Tony Cook (Manchester University) and Anton Kokalj (Jožef Stefan Institute) during their tenures as visiting scientists with CSIRO. This project was sponsored by the Advanced Materials Transformational Capability Platform (AMTCP) of CSIRO.