
Abstract
First principles thermodynamic models based on the cluster expansion formalism, lattice dynamic calculations and quantum mechanical total energy calculations are employed to compute the thermal stability of metastable hardening precipitations in hcp structure α-Mg–Gd binary alloys. It shows that vibrational entropy reverses the energetic preference and plays a critical role in hardening precipitation at different aging temperatures in Mg–Gd binary alloys. In addition to energetics, the analysis of bonding charge density reveals that the metastable β′ phase is responsible for the high strength during subsequent isothermal treatment. Our results are found to be in good agreement with experimental measurements and helpful in clarifying the metastable precipitation sequence in Mg–Gd binary alloys.
Introduction
Mg–Gd based alloy is one of the candidates for novel Mg based precipitation hardened alloys having high creep resistance and specific strength at elevated temperatures, in which the precipitates play an important role in governing their mechanical properties. However, during isothermal aging at temperatures from 20 to 500°C, the experimentally observed precipitation sequence in Mg–Gd based alloys is complex and contradictory, involving the formation of metastable phases designated β″, β′ and β1.1–8 It is initially reported that Mg–Gd binary alloys exhibit a three-stage precipitation sequence:6,7 α-Mg (SSSS)→β″ (D019 metastable)→β′ (c-bco metastable)→β (fcc stable). Then, a four-stage precipitation sequence in Mg–Gd based alloys has also been proposed. 5 5,8 The sequence consists of α-Mg (SSSS)→β″ (D019 metastable)→β′ (c-bco metastable)→β1 (fcc metastable)→β (fcc stable). The precipitation sequences mentioned above end with the stable equilibrium β. However, the detailed mechanism on how these metastable precipitates sequentially appear remains confused. Obviously, a better understanding of the precipitation process and microstructure evolution in these alloys is essential in optimising alloy design for improved Mg–Gd based alloys.
First principles calculation is a powerful tool for obtaining accurate ground state energies. Nevertheless, the inability of density functional theory (DFT) based on ab initio computation to predict accurate energy at finite temperature limits its application. Lattice dynamics calculation is one method of gaining knowledge about phonons and enables information about the system at finite temperatures to be assessed if combined with ab initio calculations. Here are two interesting examples of alloy systems where the phase boundaries were investigated by using DFT+linear response theory (LRT).9,10 One is that Ozolinš and Asta found that lattice vibrations were responsible for a 27-fold increase in the solubility of scandium in aluminium.10 In the other work, vibrational contributions were also shown to have a significant effect on the precipitation phases of the Al–Cu alloy system.11 In a landmark paper,12 Walle and Ceder gave a review on the effect of lattice vibrations on the phase stability in the alloy system. They further proposed that ab initio calculations of a complete phase diagram that include lattice vibrations could be realised by combining the cluster expansion (CE) technique with first principles thermodynamic calculations.
The current method can be summarised as follows. First, the total energies of some ordered supercells were computed for various values of lattice parameters with first principles calculations. Second, the CE technique was used to parameterise the energy of any configuration in the alloy system, and hence, some ordering structures with high probability in the actual alloy could be obtained. Finally, by comparing the free energy of the selected ground ordering structure with that of metastable precipitations, the dependence of thermal stability on temperature for these phases in Mg–Gd binary alloys was explored.
Here, we treated the solid solution ground state hcp_MgGd as well as the three reported metastable precipitation structures in Mg–Gd based alloys: β″, β′ and β1. Among them, β″ and β′ were experimentally confirmed in Mg–Gd binary alloys, while the β1 phase was reported in Mg–Gd multicomponent alloys, such as Mg–Gd–Y–Zn, Mg–Gd–Nd and Mg–Gd–Zr. However, whether β1 can precipitate in Mg–Gd binary alloys is still under debate.6,8,25 The experimental data of the equilibrium lattice parameters of the above three phases can be found in the literature.1,3 The metastable phase β″ has been identified as an ordered D019 crystal structure (hexagonal, aβ″ = 0·64 nm = 2aMg and cβ″ = 0·52nm = cMg). This phase can precipitate with perfect coherency with the matrix and can be regarded as the Mg3Gd phase in view of chemistry (Fig. 1). The intermediate β′ phase is also metastable. Its structure has a base centred orthorhombic structure (aβ′ = 0·64 nm = 2 aMg, bβ′ = 2·22 nm = 8d01Ī0Mg and cβ′ = 0·52 nm = cMg). This structure is isomorphic with the hcp structure, so the precipitation can progress simply by substituting Mg atoms with Gd without any change in the hcp lattice. The ratio of Mg/Gd is 5∶1, so it can be described as Mg5Gd. While the metastable phase β1 is a face centred based cubic. Here, we applied lattice parameters of β1 as it was in Mg–Gd multicomponent alloys, and the theoretical parameters in Mg–Gd binary alloys were obtained after global structural optimisation. The lattice parameters of β1 and α-Mg phases are assumed to be aβ1 = 0·74 nm, aMg = 0·32 nm and cMg = 0·52 nm. The orientation relationship between β1 phase and the matrix phase was such that [1 1 0] β 1//[0 0 0 1]Mg, (Ī 1 2) β 1//(1 Ī 0 0)Mg.
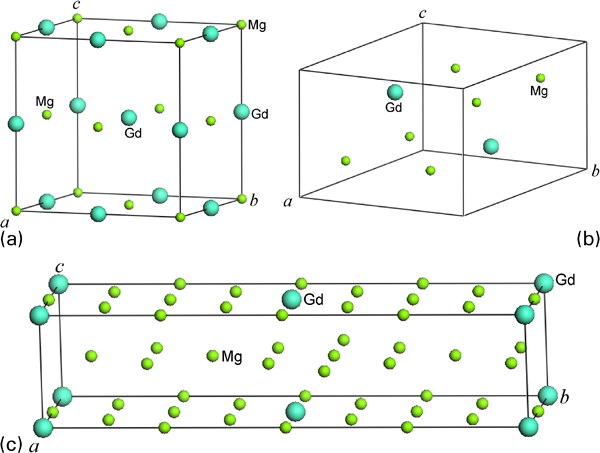
Conventional unit cell for a β1 phase, b β″ phase and c β′ phase
In the present paper, the development of microstructures involved in metastable precipitation in Mg–Gd alloys was studied using a combination of first principles total energy calculations, CE technique, and lattice dynamic calculations in the harmonic approximation. The aim of the present work is to clarify the thermal stability of precipitates during the early stages of aging precipitation in Mg–Gd binary alloys, which will be of great help in understanding the metastable precipitation sequence. In addition to energetics, the relationship between the particular microstructural changes and the alloy strength was also discussed.
Computational methods
The total energies of a large number of hcp and fcc superstructures were computed with first principles total energy calculations. Then, the formation energies ΔEform were extracted from the total energies Etot by subtracting the concentration weighted average of the total energies of the pure elements in the hcp state, i.e.
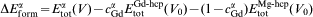
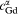 is the concentration of the Gd species in the α phase, V is the volume per atom and V0 refers to the equilibrium value of V. Nevertheless, it would be infeasible to calculate the energy of each superstructure from first principles. The CE was constructed by parameterising the formation energy of the selected atomic arrangements obtained from the above first principles calculations as a polynomial in the occupation variables σ,13 i.e.
is the concentration of the Gd species in the α phase, V is the volume per atom and V0 refers to the equilibrium value of V. Nevertheless, it would be infeasible to calculate the energy of each superstructure from first principles. The CE was constructed by parameterising the formation energy of the selected atomic arrangements obtained from the above first principles calculations as a polynomial in the occupation variables σ,13 i.e.
The DFT calculations were carried out using the ab initio total energy and molecular dynamics programme Vienna ab initio simulation package (VASP).15,16 The current calculations made use of the VASP implementation of ultrasoft pseudopotentials17 and an expansion of the electronic wave functions in plane waves with a kinetic energy cutoff of 600 eV. All the calculated results were derived by employing the generalised gradient approximation for exchange and correlation by Perdew and Wang.18 Brillouin zone integrations were performed using Monkhorst–Pack (MP) k point meshes19 and the Methfessel–Paxton technique20 with a 0·1 eV searing of the electron levels. The atomic geometry was optimised using Hellman–Feynman forces and conjugate gradients. The total energy was minimised with respect to the volume, the unit cell external degree of freedom (i.e. the unit shape) and the unit cell internal degree of freedom (i.e. the Wyckoff positions of all atoms). For each structural optimisation, tests were carried out using different k point meshes to ensure absolute convergence of the total energy with respect to the structural degrees of freedom to a precision of better than 2 meV/atom.
The remaining steps of first principles calculations were performed with the MIT ab initio phase stability software package,21 which automates most of the tasks associated with the construction of a CE. The VASP calculations were used to construct CE Hamiltonians, in which optimal cluster sets were determined by minimising the so called cross-validation score. The ECIs, which define the CE, were obtained by least squares fit to the VASP energies. The convergence criterion of the cross-validation score in our calculations was set to be <0·015 eV.
The contributions of lattice vibrations to the free energies were included on the basis of the LRT. In the harmonic approximation, the vibrational entropy as a function of temperature was calculated for ordered β′, β″ and β1 and the phase of interest representing Gd impurities in hcp Mg, such as the hcp ground state structure. In the current method, configurational and vibrational effects were considered, while the electronic free energy of electronic excitations was neglected. Below, we showed that neglecting the electronic entropy might induce an error in the calculated transition temperatures.
Results and discussion
The calculated equilibrium geometrical parameters of the three reported precipitations, β′, β″ and β1, are summarised in Table 1. In the present calculations, experimentally derived lattice parameters were used as input, and then a structure optimisation procedure was performed. Upon volume relaxation, we found that the lattice parameters and cell internal positions of the β′ and β″ phases are all in good agreement with the observations. However, the β1 structure reduced in volume by 38. It became difficult to understand how such a phase might exist in Mg–Gd binary alloys for its unreasonable lattice parameters.
Calculated equilibrium geometrical parameters and formation energies of metastable precipitations in Mg–Gd alloys: formation energies are in eV/atom
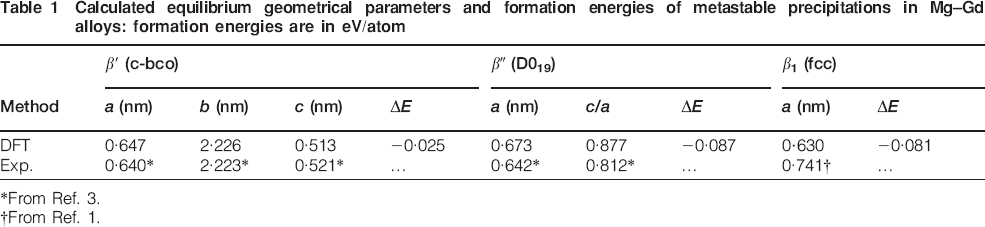
*From Ref. 3.
†From Ref. 1.
The enthalpy of formation ΔH is an important parameter for the stability of an alloy. At atmospheric pressure, the formation energy ΔEform, rather than the formation enthalpy ΔHform, could be considered. According to formalism (1), ab initio formation energies at 0 K, i.e. ΔEform, calculated with CE Hamiltonians, were plotted in Fig. 2. At zero temperature, we confirmed these results: ordering is predicted on both hcp and fcc with as lattice ground states, i.e. L3, L2, L0 and hcp_MgGd on the hcp lattice and A3 and β1 on the fcc lattice. The hcp convex hull is below that of the fcc, which implies that the hcp lattices are more stable with respect to the fcc superstructures with the same concentration. Therefore, the β1 phase, which belongs to the fcc lattice, is impossible to precipitate from hcp based Mg–Gd binary alloys at zero temperature, and its unreasonable lattice parameters can also account for this result. We found that the formation energy of β″ breaks the hcp convex hull; therefore, the β″ phase is more stable than the two phase mixtures of hcp_Mg and hcp_MgGd. However, since the formation energy of β″ (−0·087 eV/atom) is above that of hcp_MgGd (−0·1 eV/atom), β″ still cannot precipitate from Mg–Gd solid solution at 0 K. While we surprisingly found that as an experimentally observed precipitation, β′ is far above the hcp convex hall, which represents that such a configuration is not very likely to occur in the actual alloy. The first principles calculations above on the formation energies appear to contradict the precipitation of β′, β″ and β1 structures at zero temperature. In the following calculations, we investigate the contribution of lattice vibration on the free energy.
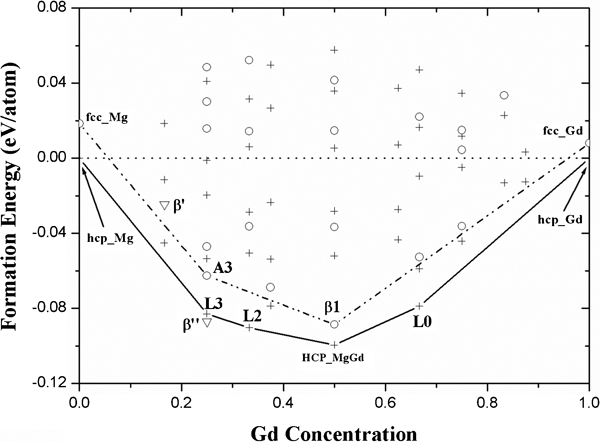
Formation energies from generalised gradient approximation calculations as defined with equation (1): superstructures of hcp (fcc) lattice are indicated with ‘+’ (‘○’), and β″ and β′ phases are indicated with ‘∇’; Convex hull pertaining to hcp (fcc) ground states is marked with solid (dash dot) line
The phase equilibria at non-zero temperature were determined with the difference of the Gibbs free energy
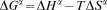
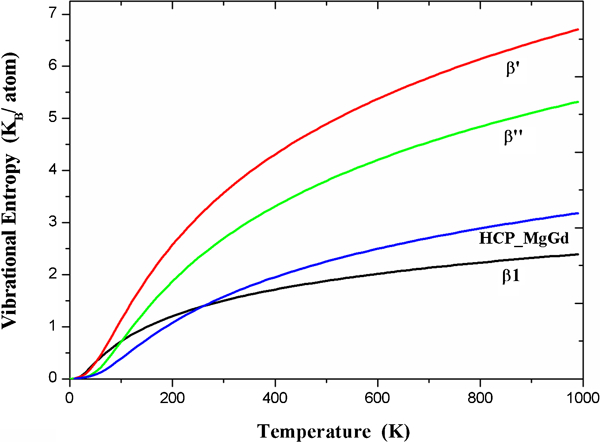
Vibrational entropies of β″, β′, β1 and hcp_MgGd from LRT calculations
On the side of the analysis of the free energy stability of precipitates, the theoretical structural transform behaviour can be elaborated as follows. At 0 K, the lattice ground state hcp_MgGd has the lowest energy, and the energy of β′ is the highest. As a consequence of the larger value of entropy of the β′ phase, the free energy of β′ descends rapidly. At the same time, the difference of the free energies between β″ and hcp_MgGd decreases with increasing temperatures because of the relatively larger vibrational entropy of β″. First, we found that β″ will be favoured over hcp_MgGd at around Tc1≈310 K. Subsequently, the free energies of β″ and β′ equal each other at a temperature of Tc2≈690 K, and the β′ phase becomes more stable. As for the β1 phase, which has the lowest vibrational entropy, it cannot precipitate during the isothermal aging of Mg–Gd binary alloys. To explain the experimental observed precipitation of β1 in Mg–Gd multicomponent alloys, we should take the effect of third party elements into account, and this topic needs to be further investigated. The details of changes of free energy for these precipitates are illustrated in Fig. 4. In the hcp_MgGd zone, the hcp_MgGd phase has the lowest free energy, and a similar explanation can be applied to the β″ and β′ zones respectively. Our calculation shows that although β″ has the lowest free energy in the β″ zone, β′ takes advantages over hcp_MgGd in terms of free energy at ∼480 K. Thus, β′ may be observed before the existence of β″ when aging at 480–690 K. When the temperature is above 690 K, β″ will convert to β′ ultimately.
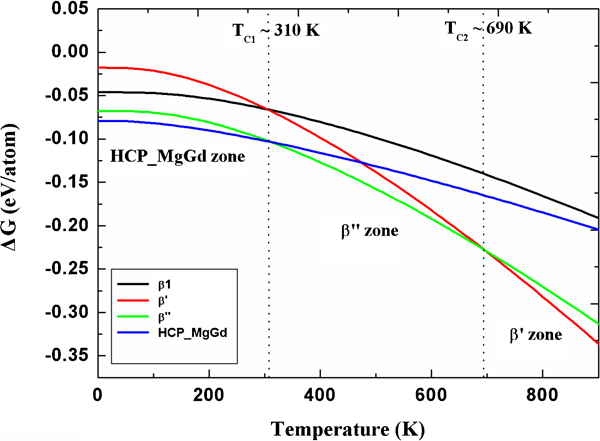
Calculated changes of free energy with respect to temperature
Microstructure evolution in isochronally heat treated Mg–Gd alloys has been investigated experimentally.4,6,7 Antion et al. characterised the aged microstructures of Mg–rare earth alloys at 423 and 523 K by TEM. They found that at the early stages of precipitation at 423 K, a D019 (β″) type ordering in the solid solution formed first, then the growth of D019 type appeared limited, while a bco (β′) structure coexisted with D019. After aging at 523 K for 4 h, the fast Fourier transform showed that the sample had the entire bco β′ structure.4 The quality of the agreement between the theoretical prediction on thermal stability and the experiment achieved for the metastable precipitations in Mg–Gd binary alloys is fairly well.
When it comes to the critical temperatures of hardening precipitations in Mg–Gd binary alloys, there is a significant scattering in transform temperatures among various measurements.1,3,4,6 So far, the exact critical precipitation temperatures of β″ and transform temperature from β″ to β′ in Mg–Gd alloys are not well determined experimentally. Therefore, the precise value of the calculated Tc should not be interpreted too strictly due to the experimental systematic errors and current theoretical drawback itself. At present, we compare our theoretical results with the commonly accepted precipitation temperature of β″ (∼423 K) and critical transform temperature from β″ to β′ (∼523 K) in Mg–Gd alloys.4 We estimated the errors due to pseudopotential and model approximation, omitting electronic excitations, and the logarithmic dependence on the vibrational frequencies results in inaccuracies of the predicted transition temperatures.
In addition, many investigations22–25 have reported that the metastable β′ phase was responsible for the peak hardness in Mg–Gd alloys during the subsequent isothermal treatment. This can be explained from our calculated bonding charge density for both β″ and β′ structures. Figure 5a displays the bonding charge density contour plot for the (001) plane of the β″ structure. The concentration of thick lines around the Gd atoms displays a build-up of charge around the Gd atoms, which is counterbalanced by a reduction in charge around the central Mg atoms, indicating that some ionicity is in action. While in Fig. 5b, which displays the bonding charge density contour plot for the (001) plane of the β′ structure, we can see clearly that a dominant feature is the concentration of charges between the Mg and Gd neighbours, indicating that covalency plays a leading role in this phase. The stronger charge bonding between Mg and Gd atoms in the β′ phase can account for its high strength.
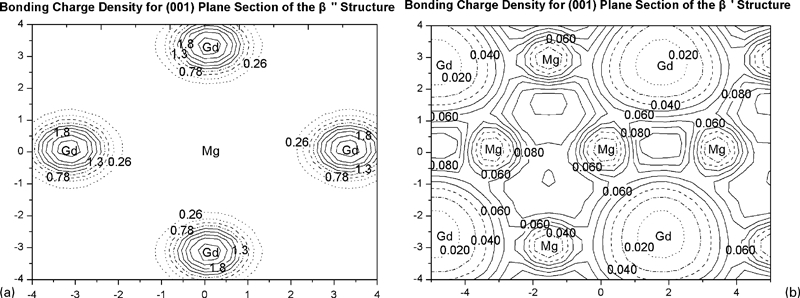
Calculated bonding charge densities for (001) plane of both β″ and β′ structures
Conclusions
The microstructure evolution at the early stages of aged precipitates of Mg–Gd binary alloys was investigated by combining the first principles free energy calculations with the CE technique. Our results showed that hcp structures are more stable than fcc, and there is no metastable precipitation except for the ordering lattice ground states at absolute zero temperature. Then, the β″(D019) structure first precipitates during isothermal aging at elevated temperature. As a consequence of the larger entropy value, the β′(c-bco) phase becomes more stable and forms at a temperature above ∼690 K. In our calculations, the precipitation of β1 (fcc) is impossible in Mg–Gd binary alloys because of its high formation energy and low vibrational entropy. The analysis of bonding charge density reveals that the metastable β′ phase is responsible for the high strength values during the subsequent isothermal treatment.
Footnotes
Acknowledgements
The authors gratefully acknowledge financial supports of the National Natural Science Foundation of China (grant no. 51001025), the Chinese Research Fund for the Doctoral Program of Higher Education (grant no. 200801451021) and the Fundamental Research Fund for the Central Universities (grant nos.N090405016 and N090502002).