
Abstract
Al2O3 nanopowders were synthesised via mechanochemical method using AlCl3 and CaO as raw materials. The effect of thermal treatment on the structural evolutions and morphological characteristics of the nanopowders was investigated using X-ray diffractometry, transmission electron microscopy, scanning electron microscopy, differential thermal analysis and Rietveld refinement. The results showed that the average crystallite size of Al2O3 was <100 nm up to ∼1200°C. The activation energy for Al2O3 nanocrystallite growth during calcinations was calculated to be ∼22 598 and 30 195 J mol−1 for η- and κ-alumina respectively, while for α-Al2O3, it was 8373 and 34 131 J mol−1 at temperatures up to 1200°C and >1200°C respectively. The mechanism of nanocrystalline growth of Al2O3 polymorphs during annealing is also discussed.
Keywords
Introduction
Aluminium oxide (Al2O3) nanopowder is a ceramic powder widely used in many fields as a single material or in multicomponent systems such as electronics, fine ceramics, composite materials, biomaterials, refractories, abrasives, catalysis, etc. The diversity in applications is due to the fact that Al2O3 has the particularity to exist in a variety of metastable structures, the so called transition alumina (such as χ, κ, γ, δ, θ and η) as well as the stable α-Al2O3,1 each having specific physic–chemical and mechanical properties. Depending on the thermal history and chemical form of the starting materials, different alumina polymorphs will be obtained.2–4 Many different processes for the production of nanopowders have been developed, such as thermal spraying, coprecipitation, sol–gel, etc.5 Precursor materials for the preparation of active alumina are usually aluminium hydroxides6–10 or sulphates.11–15
Aluminium oxide has advantages, such as its thermal, chemical and physical properties, compared with several ceramic materials. However, the surface properties and reactivity of the alumina are determined largely by their crystal structure and porous texture, which also condition the corresponding thermal behaviour during sintering or in high temperature catalytic processes.16–18
Control of microstructural and phase evolution during calcinations of alumina requires an understanding of the phase transformation sequence of the starting material.10,19–25
One of the most powerful techniques for the synthesis of a wide range of materials is a solid state process named mechanochemical treatment. This technique is a non-equilibrium solid state process in which the final product retains a very fine amorphous or nanocrystalline structure. This process can also be designed in such a way to synthesise nanocrystalline particles dispersed within a soluble salt matrix. In other words, chemical precursors react, either during milling or in the subsequent heat treatment stage, to form a nanocrystalline powder embedded within a soluble salt matrix phase. The ultrafine powder is subsequently recovered by selective removal of the matrix phase through washing with an appropriate solvent. The mechanochemical synthesis technique allows significant control over the properties of the final powder. For example, post-milling heat treatment may be used to alter the mean particle size, morphology and also the degree of crystallinity in the milled sample. Furthermore, direct formation of dispersed ultrafine particles, separated by an intervening salt matrix, suggests that agglomerate formation is more likely to be avoided as compared to other processing methods. This process is versatile, simple and cost effective and can be used to synthesise a wide variety of materials with a high yield rate on a commercial scale. The ability to decrease time and temperature of the chemical reaction at low energy consumption is another advantage of this technique.26–29
The aim of the present work is to investigate the mechanochemical treatment of anhydrous AlCl3 and CaO to obtain more information on the nature of this process and to determine the effect of thermal treatment on the structural evolutions and morphological characteristics of the nanopowder and also to obtain the activation energies of crystallite growth for the different polymorphs of alumina phases. The mechanism of nanocrystallite growth is also discussed.
Experimental
Alumina was prepared according to previously published work by the authors.30 The starting materials were anhydrous AlCl3 (Acros) and CaO (Alfa Aesar). Stoichiometric amounts of AlCl3 and CaO were used according to reaction (1) without any process control agent. The starting materials were weighted and prepared in a glove box under inert atmosphere, and after sealing the milling jar, it was transferred to the milling machine. Mechanical treatment of reactant mixtures was carried out in a FRITSCH planetary mill (planetary micromill pulverisette7) at 5000 rev min−1 under inert atmosphere using 12 and 4 alumina balls with 10 and 15 mm in diameter respectively and an alumina jar of 80 mL. Ball/powder mass ratio of 10∶1 and 5 h milling time were used in this study. All of the samples were prepared in a glove box before the milling procedure to avoid moisture adsorption.
The heat treatment in all the prepared powders was applied at a rate of 100° h−1 under air atmosphere with 1 h holding time at maximum temperature. The milled powders were annealed at 300°C, where all the starting powders had reacted. The reaction produced CaCl2 and Al(OH)3.30 The removal of the salt byproduct (CaCl2) was performed by washing the powder with distilled water several times using a sonifier and centrifuge. The washed powder was dried in an oven, and all of the subsequent heat treatments were carried out in air at temperatures between 400 and 1700°C. The phase composition and crystal structures of the prepared powders were investigated at room temperature using X-ray powder diffraction measurements. X-ray powder diffraction data were recorded at ambient conditions on a high resolution laboratory X-ray powder diffractometer [Bruker D8 ADVANCE, Cu Kα1 radiation from primary Ge(111)-Johannson type monochromator, Våntag-1 position sensitive detector in Debye–Scherrer geometry] at 40 kV and 40 mA. All the X-ray samples were filled in low absorbing glass capillaries of 0·3 mm diameter (Hilgenberg glass no. 14) and sealed in a glove box under argon atmosphere using a hot wire. Data were taken in steps of 0·008° 2θ from 5 to 90 2θ for 0·03° min−1. The samples were spun during measurement for better particle statistics. A qualitative phase analysis of the measured powder diffraction patterns using the PDF-2 database (ICCD, 2007) in combination with the program MATCH!31 was used to identify the phases. For Rietveld refinement, the Topas4-2 program32 was used. The thermal behaviour of the washed calcined as milled powder was investigated using a differential scanning analysis (DSC)/thermogravimetric (STA 409, Netzsch) analyser under static air atmosphere at a heating rate of 10°C min−1 and a sample size of ∼10 mg per run. The microstructures of the samples were analysed using transmission electron microscope (TEM; JEOL 4000 FX) and scanning electron microscope (SEM; DSM 982 GEMINI Zeiss).
Results and discussion
The starting powder mixture of AlCl3 and CaO in stoichiometric molar ratio was milled with the aim of inducing the mechanical reaction according to reaction (1)
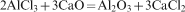
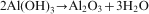
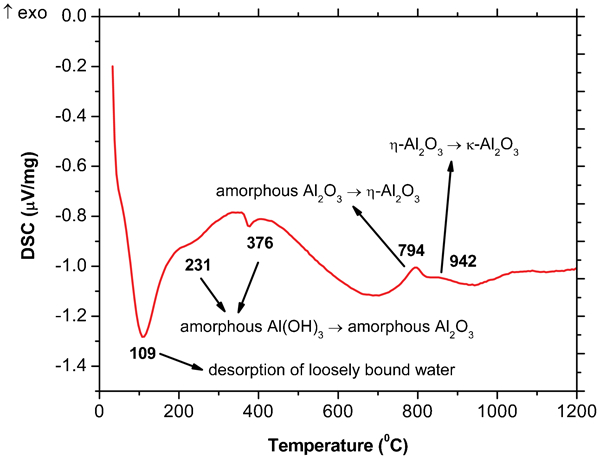
Differential scanning analysis curve of dried amorphous aluminium hydroxide powder
The amorphous aluminium hydroxide powder was calcined for 1 h at temperature ranges from 400 to 1700°C. The XRD patterns of the calcined powders at 800 and 1300°C are shown in Fig. 2. It is clear that heat treatment not only results in the transformation of the amorphous aluminium hydroxide to crystalline phases but also increases the crystallite size of the crystalline phases. Furthermore, increasing the temperature results in phase transformations as follows
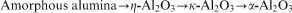
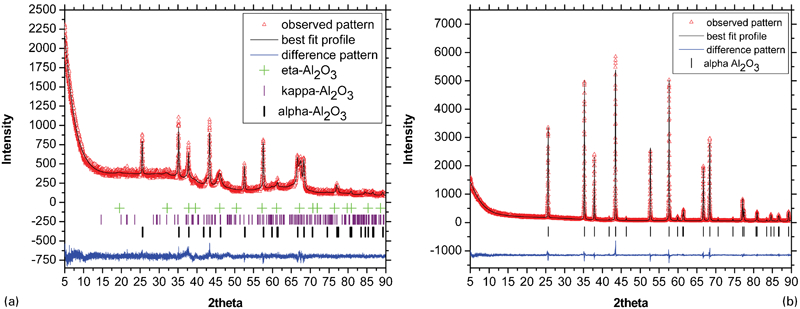
X-ray diffraction patterns of heat treated samples for 1 h at a 800°C and b 1300°C
In Table 1, the Rietveld refinement results of the X-ray patterns showing the crystallite size and weight per cent of the amorphous aluminium hydroxide samples calcined at different temperatures (400–1700°C) for the corresponding phases along with the Rwp and goodness of fit (GOF) of the Rietveld refinement are shown. According to the Rietveld refinements shown in Table 1, the phase transformation of amorphous alumina→η-Al2O3 occurs in a wide temperature range of 700–1100°C, while for the phase transformation of η-Al2O3→κ-Al2O3, it occurs in the temperature range of 800–1200°C. Increasing the temperature results in an increase in the crystallite sizes and also the phase transformations. Based on the Rietveld refinements, the η-Al2O3 phase crystallises with a size ∼7 nm from amorphous alumina at ∼700°C, and after reaching a critical crystallite size of ∼13 nm, it transforms to κ-Al2O3. The complete transformation of amorphous alumina→η-Al2O3 occurs at ∼800°C, while for η-Al2O3→κ-Al2O3, it occurs at ∼1200°C. Formation and phase transformation of transition alumina are usually exothermic and release a small amount of heat (energy). According to Table 1, the major phase transformation of amorphous alumina→η-Al2O3 occurs at ∼800°C, which is in agreement with the peak at 794°C in the DSC plot. The η-Al2O3 phase crystallises at ∼700°C with a crystallite size of 7 nm and is stable up to 1100°C with a crystallite size ∼13 nm. This phase has a critical crystallite size of 13 nm and transforms to κ-Al2O3 when the crystallite size gets larger than 13 nm. According to our Rietveld refinement for the phase transformation of η-Al2O3→κ-Al2O3, the major phase transformation occurs at ∼1000°C, which is also in a agreement with the peak at 942°C in the DSC results. Furthermore, based on Table 1, κ-Al2O3 crystallises at 800°C with a crystallite size of ∼14 nm and transforms to α-Al2O3 after reaching the critical crystallite size of ∼39 nm. The α-Al2O3 phase is the only phase present at 1300°C and higher temperatures. However, it is natural that at higher temperatures, the crystallite size of the α-Al2O3 phase is larger. Alpha alumina (α-Al2O3) is the stable form of Al2O3 where no further phase transformation will occur. As a conclusion, it should be mentioned that with the heat treatment of the amorphous aluminium hydroxide, amorphous alumina is formed and with heat treatment from 400 to 1700°C, it transforms to η-, κ- and α-Al2O3 undergoing critical crystallite size of 7, 13 and 39 nm respectively.
Crystallite size (nm) and weight per cent of aluminium hydroxide samples calcined at different temperatures for corresponding phases along with Rwp and GOF of Rietveld refinement*
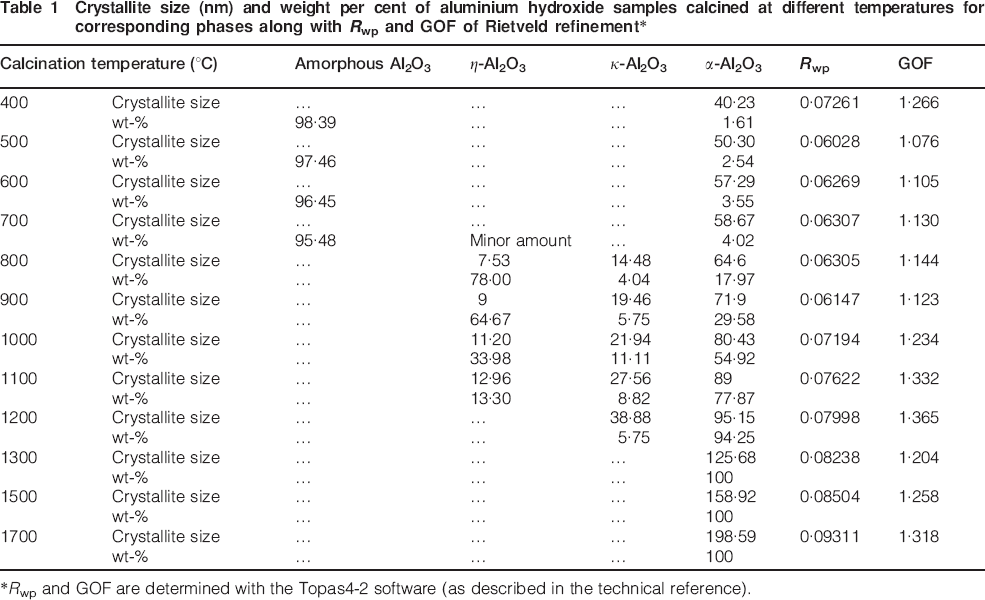
*Rwp and GOF are determined with the Topas4-2 software (as described in the technical reference).
The evolution of crystallite size of the Al2O3 powder during calcinations has been investigated, and the results obtained from Rietveld analysis of the X-ray patterns are summarised in Fig. 3.
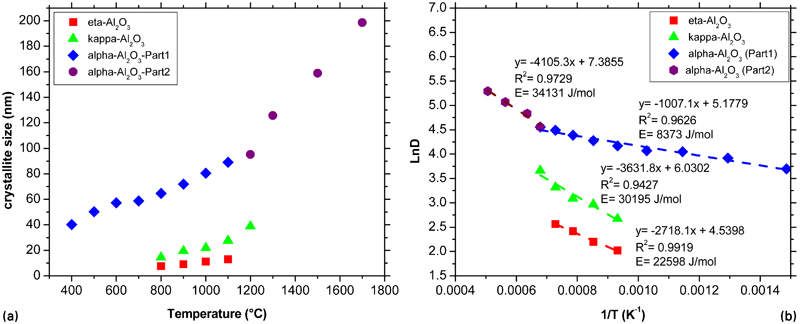
a effect of calcination temperatures on crystallite size of Al2O3 and b plot of ln D against 1/T
By increasing the temperature, the crystallite size increases. The increase in crystallite size is attributed to the typical effect of temperature on the crystal growth. It is obvious from Fig. 3a that, in the nanosize range, the nanocrystallites of different Al2O3 polymorphs grow nearly linearly, and then the rate of growth changes; this is because in the nanosize range the porosity is quite high and the pores are interconnected to maintain smaller crystal sizes,33 while for specimens with crystallite sizes higher than the nanosize range, continuous grain boundary networks have been formed due to the bridging of fine particles to increase the crystal sizes.
As shown in Fig. 3b, nanocrystallite growth during annealing can be obtained by the Scott equation by plotting the straight line of ln D against 1/T, assuming that the nanocrystallite growth is homogeneous33–36 as follows
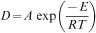

Images (SEM) of powders heat treated at different temperatures: a 600°C; b 1200°C
Figure 5 shows the TEM image of the powder calcined at 800°C. As can be seen, the size of the smaller particles is ∼5 nm, confirming that there is a wide particle size distribution. The wide particle size distribution is the main reason that phase transformations do not occur at definite temperatures and happen in a wide temperature range. As mentioned before according to the Rietveld refinements, the phase transformations occur after the crystallite size of the phases are larger than the critical crystallite size. In addition, the TEM image shows that most of the particles are spherical in shape. As mentioned before, Fig. 4 presents the SEM images of the powder heat treated at 600 and 1200°C. As can be seen in Fig. 4a, individual particles are gathered together, forming a bigger spherical particle. Figure 4b shows that the particles cling together and form larger agglomerated structures. By increasing the temperature >1200°C, the mechanism that can be activated is surface diffusion, which leads to neck formation and further agglomeration.

Image (TEM) of nanopowders heat treated at 800°C
Conclusion
Nanocrystalline Al2O3 particles have been successfully prepared by heat treatment of as milled powders obtained by mechanochemical reaction of AlCl3 and CaO as raw materials. It has been found that the phase transformation after milling, heat treatment and washing is amorphous aluminium hydroxide, amorphous alumina, eta alumina, kappa alumina and alpha alumina respectively. The calculation of the activation energy for the eta and kappa polymorphs are 22 598 and 30 195 J mol−1 respectively, growing by means of interfacial reaction. The growth mechanism for alpha alumina is interfacial reaction up to 1200°C with activation energy of 8373 J mol−1, while at temperatures higher than 1200°C its growth mechanism is grain boundary shift with activation energy of 34 131 J mol−1.
Footnotes
Acknowledgements
The authors are thankful to Professor R. Dinnebier at Max-Planck Institut für Festkörperfoschung for all his support and Professor P. A. van Aken, head of the Stuttgart Center for Electron Microscopy at Max Planck Institute for Metals Research, for the TEM experiments especially to Mr P. Kopold and Ms M. Kelsch for performing the experiments. We also extend our thanks to Mr F. Adams and Ms C. Stefani at the X-ray Service Group of Max Planck Institute for Solid State Research in Stuttgart, Germany.