
Abstract
A precursor of a zirconium diboride/silicon carbide (ZrB2/SiC) composite was synthesised via an organic–inorganic hybrid derived from gum karaya, tetraethyl orthosilicate, boric acid and zirconyl chloride starting materials. Fourier transform infrared spectroscopy of the as-synthesised dried hybrid revealed the formation of Si–O, Zr–O–C and B–O–B. X-ray diffraction revealed that the powder consists of only ZrB2 and β-SiC. Scanning electron microscopy and TEM of the composite powders showed that SiC and ZrB2 occurred in intimately mixed aggregates of spheroidal submicron sized particles for low (3M) boric acid concentration, while at high (5M) boric acid concentration, the two phases are larger with the ZrB2 adopting a blocky, angular morphology (∼10–30 μm long by 5 μm wide and thick), while the SiC remains spheroidal with ∼1 μm diameter particles in 10–20 μm diameter aggregates. Thermogravimetry–differential thermal analysis with the help of X-ray diffraction analysis revealed that the formation temperature was low at 1275°C for ZrB2 and 1350°C for the SiC with 40 wt-% yield.
Keywords
Introduction
Ultra-high-temperature ceramics based on carbides, nitrides, and borides of some transition metals have received great attention due to their potential application in extreme environmental conditions for nuclear or hypersonic aerospace uses. Such applications arise from their unique combination of material properties such as high melting point (>3000°C), high thermal and electrical conductivities, chemical inertness and excellent ablation/oxidation resistance. In particular, zirconium diboride (ZrB2) has attracted much attention.1–11
Silicon carbide (SiC) is a common additive to promote densification and oxidation resistance of ultra-high-temperature ceramics.9,10 It is frequently used in powder form or as thin layers in many industrial applications such as wear-resistant coatings, corrosion or oxidation protection barriers.12,14 Conversely, the poor oxidation resistance of ZrB2 at elevated temperatures limits its use in many potential applications. Silicon carbide is known to provide passive oxidation protection when added to ZrB2 through intermediate temperature ( < 1800°C) regimes. 14
Conventional synthesis of ZrB2 via powder mixing, ball milling and solid state sintering (in an SPS) often leads to contamination of the final product from the process, and attrition of milling media used increases the content of impurities that may also be inhomogeneous because the powders are mixed together on a relatively coarse scale (e.g., micrometre scale). As a result, in the present work, an organometallic preceramic polymer precursor method was used, which is regarded as an effective way for the low temperature synthesis of homogeneous ultra-fine ceramic powders.
The objective and novelty of the present work is to synthesise composites of zirconium diboride/silicon carbide (ZrB2/SiC) from an organic–inorganic polymer precursor route by boro/carbothermic reduction. By using the precursors and controlling the synthesis conditions, it will be possible to prepare homogeneous and pure biphasic ceramic composites that have the advantages of lower reaction temperature and shorter reaction time due to the intimate mixing of the reactants.
Experimental
The starting materials were zirconium oxychloride octahydrate [ZrOCl2·8H2O (ZOO)], gum karaya (GK), boric acid [H3BO3 (BO)], and tetraethyl orthosilicate (TEOS) with a purity of ≥ 99.5%, and all were supplied by Sigma-Aldrich (Dorset, UK).
Figure 1 is a flowchart for the synthesis of ZrB2/SiC, illustrating that the solutions containing zirconium, boron and silicon were prepared separately. The carbon, zirconium and boron containing solutions were prepared by dissolving GK in water at 60°C before cooling to room temperature. Additions of ZOO and boric acid were then made to the solution under continuous stirring. Molar concentration of boric acid was varied (2, 3, 4 and 5M) to study the influence on the purity of the final powder. On the other hand, a carbon and silicon based solution was prepared by dissolving GK in water at 60°C. After cooling, TEOS was added to the GK solution with constant stirring until a homogeneous solution was formed. The two solutions were mixed together adding the GK/zirconium/boron solution to the GK/silicon solution under continuous stirring. The solution was dried at 110°C to remove the solvent. The dried gel was heat treated in an alumina boat in an alumina tube furnace (Lenton Furnaces and Ovens, Derbyshire, UK) at 1400°C for 3 h in flowing argon (60 mL min− 1) at a heating rate of 10°C min− 1.
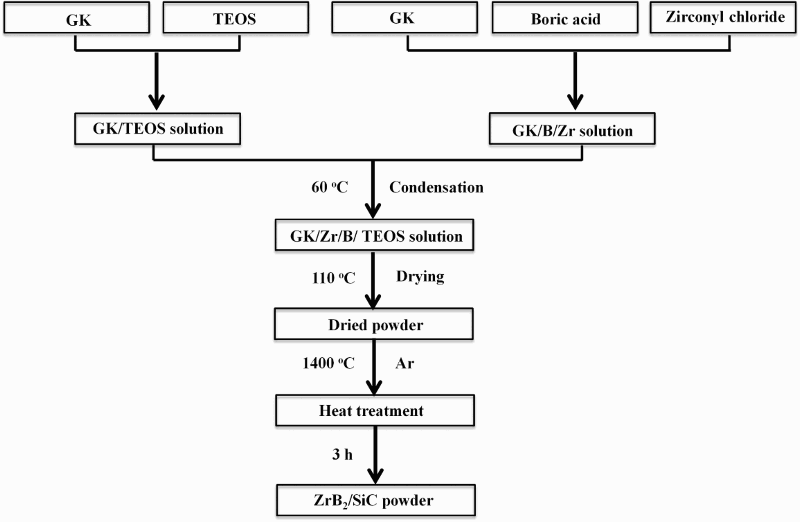
Flowchart for synthesis of ZrB2/SiC
Zirconium diboride powder can be prepared by the following synthesis routes: carbothermal reduction [chemical reaction (1) (Refs. 10 and 15], borothermal reduction [chemical reaction(2) (Refs. 16 and 17)], and via a combined boro/carbothermal reduction method [chemical reaction (3) (Ref. 18)]
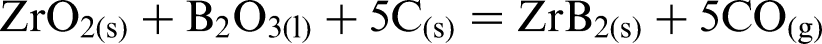
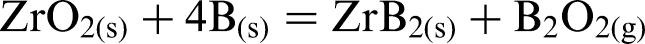
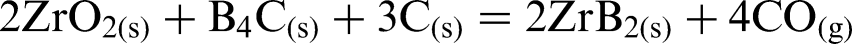
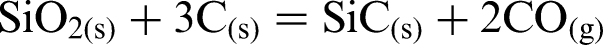
X-ray diffraction (XRD) (PANAlytical Xpert 3 diffractometer, Cambridge, UK) with Cu Kα1 radiation (λ = 1.540598 Å) and a secondary monochromator scanned from 20° to 90° 2θ was used to identify the phase composition of the synthesised powders. The crystallite size was calculated from the XRD diffractogram based on the Scherrer equation. X-ray line broadening for all peaks corresponding to zirconium diboride as well as silicon carbide was taken into consideration when performing this calculation. The morphology of the phases in the composite powders was investigated using secondary electron imaging in a scanning electron microscope (SEM, JEOL JSM 6010 LA, Tokyo, Japan) equipped with energy dispersive X-ray spectroscopy (EDX). A transmission electron microscopy (TEM, JEOL JEM 2000FX, Tokyo, Japan) with an acceleration voltage up to 200 kV was used to analyse the morphology of the fine particles using bright-field imaging. Fourier transform infrared spectroscopy (FT-IR) spectra of as-synthesised composite precursors were acquired using a Nicolet iS10 spectrometer (Thermo Scientific Company Ltd, Waltham, USA) in the range of 4000–400 cm− 1 to identify structural changes and interactions of organic ligands with the metal salt ions. All spectra were baseline corrected and normalised thereafter to the highest peak. Thermal analysis [thermogravimetry–differential thermal analysis, (TG–DTA)] of the as-synthesised powder dried at 110°C was performed at a heating rate of 10°C min− 1 in the range of 30 to 1550°C under argon flow (60 mL min− 1) using NETZSCH STA F1 Jupiter (Selb, Germany).
Results
Figure 2 shows XRD of synthesised composite powders heat treated at different temperatures to identify the formation of ZrB2/SiC (phase compositions and crystalline state of the composite powders). After heat treatment at 1200°C, t-ZrO2 and m-ZrO2 were detected. No B2O3, SiO2 and C were detected, indicating that these phases were present in amorphous form or in amounts too small to be detected by XRD. Initial formation of ZrB2 was observed at ∼1300°C along with some monoclinic zirconia (m-ZrO2). After heating the precursor at 1400°C, ZrB2 and SiC were detected, but ZrO2 was not.
X-ray diffraction of synthesised composite powders heat treated for 3 h at 1200, 1300, 1400 and 1500°C under Ar
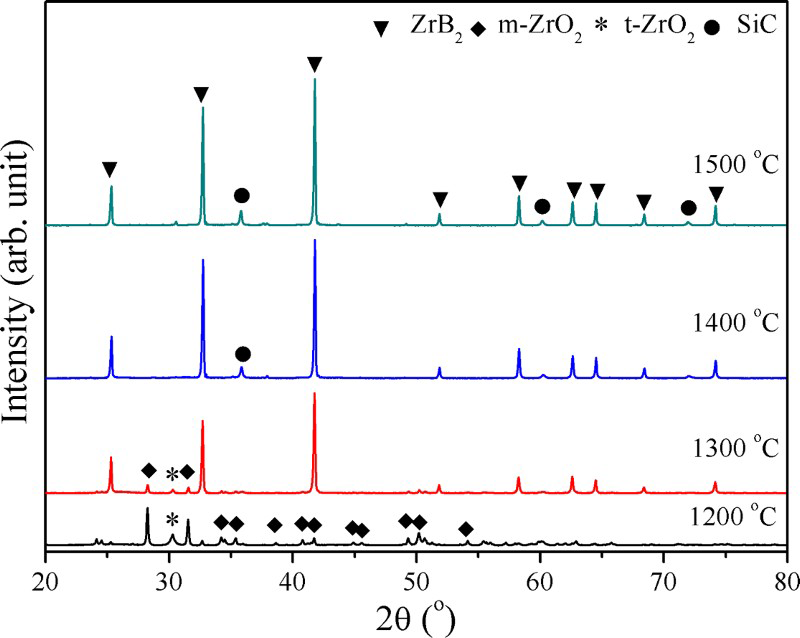
Figure 3 shows the influence of boric acid concentration on the boro/carbothermal reduction and hence phase composition of the as-synthesised dried powder after 3 h at 1400°C in argon. X-ray diffraction from the ZrB2/SiC composites made using 2M (B2M) includes significant amounts of ZrC impurity, whereas at 3M (B3M) boric acid concentration only ZrB2/SiC peaks occur. This confirms the need to include excess boric acid in the initial mix to allow for B2O3 losses by vaporisation. 24 On further increase in boric acid concentration to B4M and B5M, there is no change except for increased intensity of the XRD peaks.
X-ray diffraction of synthesised composite powders made with varying boric acid concentrations (B2M, B3M, B4M and B5M) heat treated for 3 h at 1400°C under Ar
Fourier transform infrared spectroscopy has been used to investigate the structural changes in materials by various investigators.25–27 Figure 4a and b shows the normalised FT-IR absorption spectral band of starting materials used for synthesis of the hybrid precursor, i.e. GK, ZOO, BO, TEOS and the hybrid complex after drying at 110°C to determine the structural changes and interactions of organic ligands with the metal salt ions (e.g. Zr, B and Si). For GK (Fig. 4a), the major bands observed correspond to vibrations of characteristic groups; the broad band at 3330 cm− 1 corresponds to the O–H stretching band of the hydroxyl group; the band at 1725 cm− 1 is due to the C = O stretching vibration, that at 1595 cm− 1 to the C = C band of
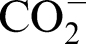 asymmetric stretching from carboxylic acid salts, that at 1417 cm− 1 to the
asymmetric stretching from carboxylic acid salts, that at 1417 cm− 1 to the
 symmetric stretching and 1371 cm− 1 represents the C–H deformation. The band at 1244 cm− 1 is due to the
symmetric stretching and 1371 cm− 1 represents the C–H deformation. The band at 1244 cm− 1 is due to the
 symmetrical stretching from carboxylic acid salts or C–O–C asymmetrical stretching. For ZOO (Fig. 4a), the band at 1623 cm− 1 can be assigned to the ‘scissor’ bending mode of coordinated water. The wide peak at 3330 cm− 1 can be attributed to the stretching vibration of the –OH group. For BO (Fig. 4a), the characteristic O–H stretching band of the hydroxyl group appears at 3183 cm− 1. The absorption band near 1410 cm− 1 was assigned to the antisymmetric stretching vibration of the B–O band. The intense peak at 545 cm− 1 was attributed to the vibration of the B–O band. For TEOS (Fig. 4a), no O–H stretching band was observed as it is free from water. The intense peak at 1073 cm− 1 corresponds to Si–O–C bond or the Si–O–Si bond. The band observed at 465 cm− 1 could be assigned to the Si–O–Si bond. The major band at 783 cm− 1 is related to the Si–O band. For the as-synthesised hybrid complex (Fig. 4b), the O–H stretching band appears at 3220 cm− 1 with three small shoulders. The intensity of the bands at 1725 and 1595 cm− 1 was almost diminished. The band at 1335 cm− 1 was intense. In addition, there were several new peaks presumably arising from the hybrid complex.
symmetrical stretching from carboxylic acid salts or C–O–C asymmetrical stretching. For ZOO (Fig. 4a), the band at 1623 cm− 1 can be assigned to the ‘scissor’ bending mode of coordinated water. The wide peak at 3330 cm− 1 can be attributed to the stretching vibration of the –OH group. For BO (Fig. 4a), the characteristic O–H stretching band of the hydroxyl group appears at 3183 cm− 1. The absorption band near 1410 cm− 1 was assigned to the antisymmetric stretching vibration of the B–O band. The intense peak at 545 cm− 1 was attributed to the vibration of the B–O band. For TEOS (Fig. 4a), no O–H stretching band was observed as it is free from water. The intense peak at 1073 cm− 1 corresponds to Si–O–C bond or the Si–O–Si bond. The band observed at 465 cm− 1 could be assigned to the Si–O–Si bond. The major band at 783 cm− 1 is related to the Si–O band. For the as-synthesised hybrid complex (Fig. 4b), the O–H stretching band appears at 3220 cm− 1 with three small shoulders. The intensity of the bands at 1725 and 1595 cm− 1 was almost diminished. The band at 1335 cm− 1 was intense. In addition, there were several new peaks presumably arising from the hybrid complex.
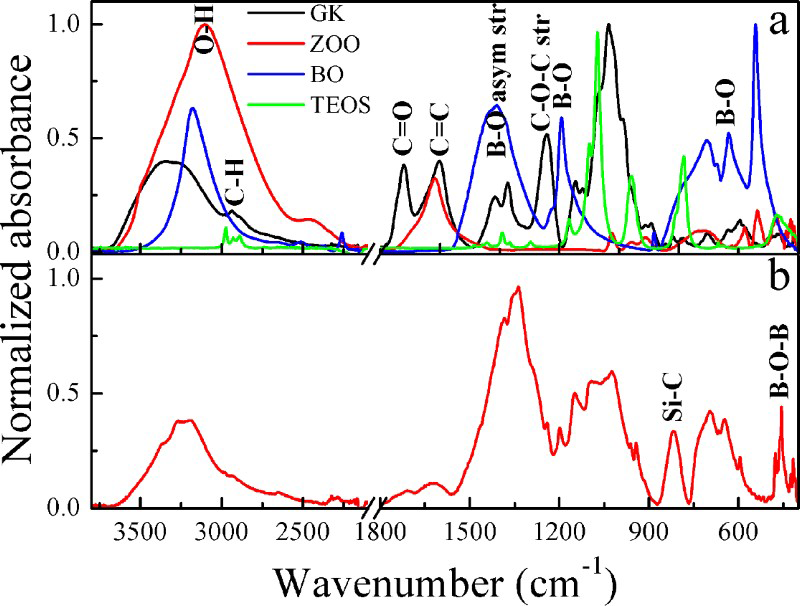
Fourier transform infrared spectroscopy of a starting materials and b as-synthesised sample dried at 110°C
A representative secondary electron SEM image of the composite powder synthesised using 3M boric acid is shown in Fig. 5a revealing formation of spheroidal particle aggregates. The particle size and morphology were observed to depend on the synthesis composition and reaction condition. The X-ray dot mapping shows reasonably uniform distribution of the elements, suggesting that the SiC and ZrB2 are intimately mixed. On the other hand, larger, blocky, angular features formed when the boric acid concentration was above 3M concentrations as shown in Fig. 6. X-ray dot maps of the angular features reveal that they are ZrB2. Hence, at high (5M) boric acid concentration, the two phases are larger with the ZrB2 adopting a blocky, angular morphology (∼10–30 μm long by 5 μm wide and thick), while the SiC remains spheroidal with ∼1 μm diameter aggregates.
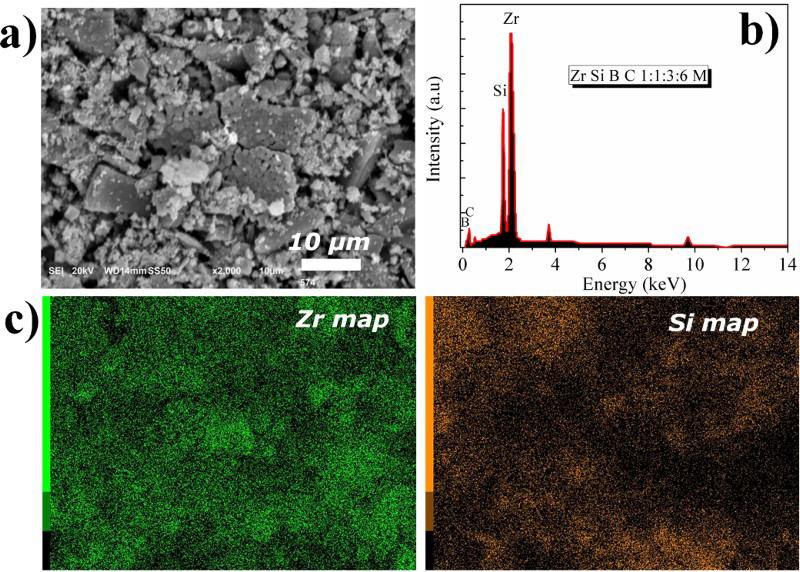
a Micrographs (SEM), b EDX spectra and c X-ray dot mapping of composite powder heat treated for 3 h at 1400°C for 3M boric concentration

a Micrographs (SEM) and b and c EDX dot mapping of the composite powder heat treated for 3 h at 1400°C for 5M boric concentration
Transmission electron microscopy imaging of the synthesised powder (Fig. 7) revealed two different particle morphologies confirming the SEM observations of intimately mixed small submicron particles of SiC and ZrB2 with 3M boric acid and blocky, angular ZrB2 with 5M boric acid. Transmission electron microscopy investigation of the powders revealed no evidence of the presence of amorphous carbon consistent with the results obtained from the XRD that the synthesised sample is composed of ZrB2/SiC crystals only.

Bright-field TEM micrographs of composite powder synthesised using a 3M boric acid and b 5M boric acid both heat treated for 3 h at 1400°C under Ar
Thermogravimetric analysis has been used to investigate the decomposition, stability, purity and yield by various investigators.28–31 Figure 8 shows representative TG and DTA curves of dried samples as-synthesised under argon atmosphere. Thermogravimetry reveals that the mass loss for the hybrid occurs in three steps with respect to the temperature. The first major mass loss (40%) occurs from 170 to 700°C with second mass loss (3%) from 700 to 1275°C. The third mass loss (27%) occurs from 1275 to 1550°C. Differential thermal analysis of the hybrid complex in argon shows four major reactions at 150, 200, 570 and 1275°C.
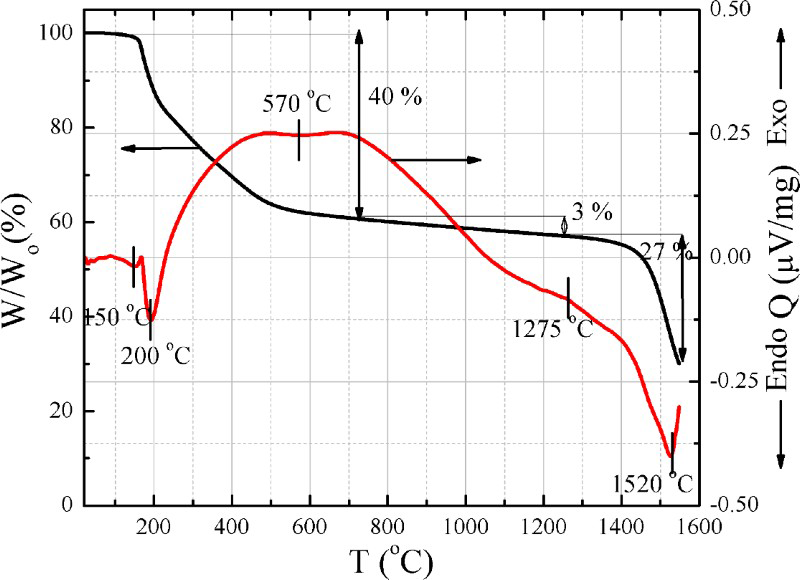
Thermogravimetry (W/Wo represents mass loss) and DTA (Q represents heat flow) of as-synthesised hybrid precursor in argon
Discussion
X-ray diffraction of synthesised composite powders heat treated for 3 h at 1400°C under argon (Fig. 2) revealed sharp peaks with the absence of any humps indicative of amorphous phases suggesting that the reaction between the starting precursors has led to complete formation of fully crystalline phases. Furthermore, the absence of any extra peaks in the pattern for 1400°C (Fig. 2) indicates that the synthesised powder consisted of a composite of zirconium diboride and silicon carbide. X-ray diffraction reveals that a composite of ZrB2/SiC free of any impurities is produced at a relatively low reaction temperature of 1400°C. This temperature of formation is much lower than that used for preparing ZrB2 alone via solid state synthesis, which is 2000°C. 32
X-ray diffraction of synthesised composite powder by varying boric acid concentration heat treated for 3 h at 1400°C under argon (Fig. 3) revealed a small amount of cubic ZrC as impurity for 2M (B2M) boric acid indicating that the amount of boron is insufficient (since some B2O3 vaporised at high temperature) for the reaction, which led to the formation of ZrC. On increasing the boric acid concentration to 3M (B3M), no ZrC impurity was found. Therefore, boric acid must be used in excess to compensate the B2O3 loss at high temperature.
Comparing the FT-IR results of the starting materials with those of the as-synthesised dried hybrid precursor reveals that the structural changes occur. The shoulders appearing in the O–H band for the as-synthesised hybrid complex (Fig. 4b) are an indication of the association of Zr, B and Si with the organic polymer. The peaks at 695 and 645 cm− 1 were attributed to B–O bond vibrations. The absorption band at 1340 cm− 1 was assigned to the antisymmetric stretching vibration of the B–O bond, which confirms the condensation reaction of GK with boric acid. The existence of a B–O–B linkage was indicated by the peak located at 495 cm− 1. The organic ligands ν(C = O) chelate to zirconium, which is evident from the differences in the FT-IR spectra. The peaks at 1723 and 1604 cm− 1 assigned to ν(C = O) of GK indicate that it chelates to the zirconium ion that is shifted to a lower wave number of 1711 cm− 1 forming Zr–O–C. The B–O bond of boric acid chelates to the Zr–O–C that induced condensation, thus forming a Zr–O–C–B complex network evident from the band shifting to lower wave numbers (1385 and 1338 cm− 1). An intense peak in the spectra of the as-synthesised hybrid was found at 817 cm− 1. This peak was assigned to a Si–C stretching vibration band that is significantly higher than the previously recorded value. 33 This shift may be due to the size of the particle being too small to be comparable to the wavelength of the electron leading to a transition from continuous to discrete energy levels that makes the Si–C band shift to a higher wave number resulting in a blue shift. 34 Furthermore, the shift to a higher wave number of the Si–C band was attributed to the fact that when the crystallites of SiC are larger than micrometre size, the peak position is close to 800 cm− 1. However, for nanosized crystallites, the frequency is higher than the micrometre size. 35 This is consistent with the calculated SiC crystallite size of 40 ± 4 nm.
Depending on the boric acid concentration during synthesis, the resulting ZrB2/SiC powder had different morphologies. Using 3M boric acid led to an intimate mix of aggregated submicron spheroidal SiC and ZrB2 particles (Figs. 5 and 7a) in which EDX indicated uniform composition but XRD indicated that it was biphasic, whereas the ZrB2 (confirmed by EDX) had a blocky, angular morphology when high boric acid (5M) concentration was used (Figs. 6 and 7b). At 1400°C, for low boric acid concentration (3M), the liquid B2O3 in the system was consumed both by the reaction and evaporation, while at high boric acid concentration (5M) the liquid B2O3 phase was still present in excess. With extra liquid B2O3 at 1400°C, the blocky morphology of the ZrB2 grains was more prominent compared to 3M boric acid concentration. The liquid B2O3 surrounds and directly contacts the ZrB2 clusters enabling them to coalesce and grow in preferred crystallographic directions. As a result, the morphology of the ZrB2 powders showed blocky structure at high boric acid concentration.
From the TG analysis, it is evident that there is a slight change in mass (1%) at 150°C due to adsorbed moisture. The change in mass (13%) at 200°C is likely due to the dehydration of boric acid to form B2O3. 36 This B2O3 is in amorphous form, which does not have a true melting point. Amorphous B2O3 is liquid above 500°C explaining the onset of a small melting peak in the DTA at 500°C. After 200°C, the organic polymer (GK) used in the complex decomposed to form carbon. The total mass loss at 700°C is ∼40%. The small mass changes (3%) from 700 to 1275°C are due to the evaporation of some foreign materials from the organic–inorganic moieties during the polymorphic changes of oxides (e.g. ZrO2, B2O3 and SiO2). The second major mass loss starting at ∼1275°C is due to initiation of the boro/carbothermic reaction [equation (3)] followed by evaporation of CO. From DTA of the hybrid complex, four prominent reactions occur. The first two endothermic peaks at 150 and 200°C are due to the evaporation of water and dehydration of the boric acid to form B2O3. 36 Decomposition of the organic polymer shows a broad exothermic peak. A small endothermic shoulder was observed at ∼570°C, which is likely due to the melting of B2O3. The boro/carbothermic reaction started at ∼1275°C with a clear endo peak, which is an indication of the formation of ZrB2, while for SiC it was at 1400°C.
B2O3 had dissociated into B during the course of the ZrB2 formation in the precursor or the amorphous B had oxidised into B2O3. As the reaction was carried out in argon atmosphere in the presence of carbon, the likelihood for the latter is extremely small. This result indicates that the formation of ZrB2 at ∼1300°C most likely occurs through formation of amorphous boron or boron suboxides as the intermediate product in chemical reactions (2) and (3), although at higher temperatures there could be more than one reaction mechanism taking place simultaneously. B2O3 has an unusually low melting point (500°C) and a high vapour pressure. The vapour pressure of B2O3 at 1527°C reaches 344 Pa, leading to its rapid vaporisation. 16 On using 2M boric acid (B2M), minor levels of ZrC impurity are detected (Fig. 3), which as described previously is insufficient to replace all carbon due to evaporation of B2O3 so that extra boron must be added to compensate the loss of boron oxide. On using 3M boric acid (B3M), no ZrC appeared. The ZrB2/SiC composites obtained at 1400°C showed a blocky, rod-like morphology for ZrB2 when the molar concentration of boric acid is 5, whereas the ZrB2/SiC obtained at 1400°C for 3M boric acid concentration have a spheroidal morphology. The B2O3 (liquid) phase exists in the system when the synthesising temperature was above 1000°C and started to evaporate above 1100°C forming gaseous boron oxide as revealed by Ito et al. 37 The blocky morphology of ZrB2 in the 5M boric acid system may be attributed to the presence of liquid B2O3. At 1400°C, for low boric acid concentration (3M) during the whole duration of dwell time, the liquid B2O3 in the system was consumed both by the reaction and evaporation, while at high boric acid concentration (5M) the liquid B2O3 phase was still present in excess. Therefore, the change in ZrB2 crystal shape is likely due to the presence of the liquid phase. With additional liquid B2O3 at 1400°C, the ZrB2 crystals were able to assume the angular, blocky habit. The liquid B2O3 contact with the ZrB2 clusters enables their free growth and adoption of a characteristic crystal habit not formed with less liquid present.
Conclusions
A solution based route using an organic–inorganic hybrid containing Zr, B and Si was used to prepare homogeneous and pure biphasic ceramic composite powders. The crystallite size of the composite powder was found to be ∼80/40 ± 5 nm for ZrB2/SiC phases with the absence of any residual carbon for the optimised condition of a Zr/Si/B/C ratio of 1:1:3:6. An impurity of zirconium carbide was found to exist in the synthesised powder while using a 2M boric acid concentration. The synthesis temperature (1400°C) for this process is suitable to form ZrB2/SiC with a small dwell time of 3 h, which is much lower than the conventional solid state process. Boro/carbothermal reduction temperatures as low as 1400°C could be used to form ZrB2/SiC as revealed by thermogravimetric analysis. Scanning electron microscopy reveals that spherical particles for 3M boric acid concentration due to liquid B2O3 in the system were consumed both by boro/carbothermic reduction and evaporation, while for 5M boric acid concentration liquid B2O3 remains in significant quantities providing mass transport and allowing the crystals to grow large and with angular habit.
Footnotes
Acknowledgements
The financial support provided by the Office of Naval Research Global, USA under contract number N62909-13-1-N055 is gratefully acknowledged.