
Abstract
We present a 3-month-old girl who displayed typical clinical characteristics of blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES). She was referred to our clinic with an initial diagnosis of Down syndrome. Clinical features of elevated follicle stimulating hormone and low estradiol levels in the case were diagnosed as BPES syndrome and were consistent with BPES type 2. To date, there are no cases of BPES with cleft palate and cardiomyopathy, suggesting that these novel findings can be part of this condition.
The blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES; also termed Waardenburg syndrome) was first reported by Vignes in 1889. BPES is a rare disease characterized by blepharophimosis, ptosis, epicanthus inversus, and telecanthus, which are generally inherited in an autosomal dominant manner (Beaconsfield et al., 1991). Its prevalence is approximately 1:50,000. In BPES, non-ocular associations include broad-flat nasal bridge, high-arched palate, protruding or cup-shaped ears, female infertility (ovarian failure), and cardiac defects (De Baere et al., 2001; Beckingsale et al., 2003; Choi et al., 2006). There are two types of this condition: eyelid malformations with amenorrhea and/or premature ovarian failure characterized by lower estrogen and elevated gonadotropin levels in type 1 and no ovarian failure in type 2 (Crisponi et al., 2001; Méduri, 2010). Hypertrophic cardiomyopathy and cleft palate are the novel findings in our case diagnosed as BPES by characteristics clinical findings, which have not been reported so far. We emphasize two novel findings in this rarely seen syndrome by presenting this case that is consistent with BPES type 2. In addition, BPES has not yet been reported in Turkey.
Case Report
A 3-month-old girl was referred to a pediatric endocrinology outpatient clinic with an initial diagnosis of Down syndrome. Prenatal follow-up was normal. She is the second child of a mother aged 28 years and a father aged 35 years. She was born via normal spontaneous vaginal delivery with a birth weight of 3100 g. She was admitted to a neonatal unit for 15 days due to a cleft palate. At presentation, body weight was 3800 g (<3rd percentile) and height and head circumference were 57 cm (<3rd percentile) and 38 cm (3rd percentile), respectively. Anterior fontanel was found to be normal. On physical examination, there were blepharophimosis, ptosis, epicanthus inversus, telecanthus, microphthalmia (Fig. 1), microcephaly, cleft palate, low-set ears (Fig. 2), widely spaced nipples, and growth and neurodevelopmental retardation. There was no other abnormal finding on physical examination. Thyroid function was found to be normal, and follicle stimulating hormone (FSH), luteinizing hormone, and estradiol levels were 28.9 mIU/L, 0.76 IU/L, and <10 pg/mL, respectively. These values were compatible with ovarian failure at the minipuberty period. Complete blood count and liver and renal function tests were normal, and karyotype was 46, XX. No additional anomaly was detected on renal sonography and bone radiographs. Hypertrophic cardiomyopathy was detected on echocardiography. No abnormality other than cleft palate was detected on brain magnetic resonance imaging (Fig. 3).
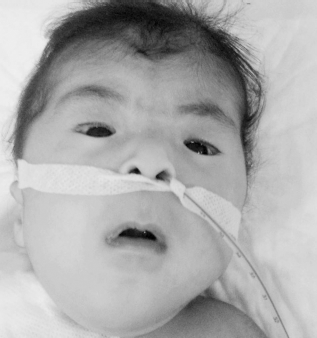
Image of blepharophimosis, ptosis, epicanthus inversus, microphthalmia, and telecanthus.
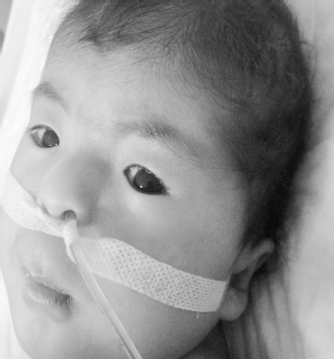
Image of blepharophimosis, ptosis, epicanthus inversus, microphthalmia, telecanthus, microcephaly, and a low-set ear.

Cleft palate in the (A) sagittal magnetic resonance image and (B) axial magnetic resonance image.
Discussion
The primary syndromes accompanied by blepharophimosis include Marden-Walker syndrome, Ohdo syndrome, and Michels syndrome. Marden-Walker syndrome is characterized by multiple joint contractures, a mask-like face with blepharophimosis, micrognathia, high-arched or cleft palate, low-set ears, arachnodactyly, kyphoscoliosis, decreased muscular bulk, failure to thrive, and marked motor and mental retardation (Begum and Nayek, 2002). In Marden-Walker syndrome, no ocular finding other than blepharophimosis such as ptosis, epicanthus inversus, and telecanthus are present, but severe joint contractures and abnormalities of the central nervous system on sonography and magnetic resonance imaging studies can be observed. Camptodactyly, chest deformation as pectus (excavates or carinatus), kyphoscoliosis, and absent deep tendon reflexes are common (Ben-Neriah et al., 1995). In our case, normal brain imaging studies and a lack of skeletal system abnormalities facilitated differentiation from Marden-Walker syndrome. Ohdo syndrome (OS) is associated with blepharophimosis and blepharoptosis as well as typical facial features, including a round face with a characteristic nose and a narrow mouth (Verloes et al., 2006). In addition, epicanthus inversus and telecanthus have not been reported in OS. Moreover, thyroid abnormality can be seen in OS. Although cryptorchidism and scrotal hypoplasia have been reported in OS, hypergonadotrophic hypogonadism was present in our case, and this is not among findings reported with OS. A diagnosis of OS was readily excluded by these discriminative findings, especially in the presence of a facial appearance that is characteristic to BPES. Blepharophimosis, blepharoptosis, and epicanthus inversus (BBE), the classical triad of Michels syndrome, were all present, but radioulnar synostosis and anterior chamber defects that are reported in a majority of Michels syndrome cases were lacking in our patient. Moreover, telecanthus and highly arched brows are seen in up to 80% of Michels syndrome cases (Online Mendelian Inheritance in Man, 2007). Telecantus and low-set ears in our case are among findings reported with Michels syndrome; however, highly arched brows, skeletal abnormalities, and abdominal diastasis were lacking in our case. In addition, all 11 Michels syndrome cases reported to date consistently exhibited the BBE triad plus a cleft lip and palate (Adedayo et al., 2014). In our case, a lack of cleft lip in addition to the above-mentioned findings that are inconsistent with Michels syndrome excluded a Michels syndrome diagnosis.
All four eyelid findings (blepharophimosis, ptosis, epicanthus inversus, and telecanthus), which are characteristics of BPES, were present in our case. Again, broad-flat nasal bridge, low-set ear, and microphthalmia in our case were also among reported characteristics of BPES (Beckingsale et al., 2003; Choi et al., 2006), thus supporting a BPES diagnosis. Elevated FSH and low estradiol levels in a case diagnosed as BPES syndrome by clinical features were consistent with BPES type 2, which is accompanied with ovarian failure. However, cleft palate is not among reported characteristics of BPES. When conditions involving an association of cleft palate with facial dysmorphism were reviewed in the literature, trisomies were excluded in our case due to a karyotype of 46, XX. When conditions involving an association of cleft palate with facial characteristics of BPES were reviewed, it was determined that hereditary neuralgic amyotrophy (HNA) should be discriminated in our case. Cleft palate and dysmorphic facial characteristics have been previously identified in classic HNA cases (Erikson, 1974; Airaksinen et al., 1985; Orstavik et al., 1997; Jeannet et al. 2001). Although there are a few reports on the facial aspect of patients with HNA, almost none affect individuals in early childhood (Dunn et al., 1978; Airaksinen et al., 1985; Orstavik et al., 1997; Jeannet et al., 2001; Laccone et al., 2008). In a case report, an HNA diagnosis was made in a patient in early childhood based on a history of repetitive unilateral brachial neuritis in the patient's father and paternal grandmother (Laccone et al., 2008). In our case, an HNA diagnosis was excluded due to a lack of symptoms suggestive of HNA.
Cardiac defects have been reported in BPES; however, data on comprehensive assessments of cardiac findings are limited. To the best of our knowledge, hypertrophic cardiomyopathy has not been reported. When conditions involving an association of cleft palate with hypertrophic cardiomyopathy were reviewed, it was found that there was a single case with proximal trisomy of 1q mosaicism where hypertrophic cardiomyopathy was associated with micrognathia, cleft palate, low-set ears, posterior localization of thumbs, and bilateral syndactyly of the second and third toes (Hirshfeld et al., 2001). However, in our case trisomy was excluded due to a karyotype of 46, XX. As a result of these considerations, our case was found to be consistent with BPES type 2 given the typical clinical characteristics and hormone profile that indicates ovarian failure.
Cleft palate and hypertrophic cardiomyopathy can be part of this condition. Given the fact that BPES has genetic heterogeneity, these novel findings are not surprising. For example, a hypergonadotropic pattern is a characteristic feature in ovarian failure seen in BPES but has been reported in only one case (Schlade-Bartusiak et al., 2012). Again, two BPES cases with congenital alacrima were reported in the literature. Among these, there was congenital deficiency of the lacrimal gland without sweat production in one case, but it was normally developed in the other case (Ng et al., 2014). Mutation in the FOXL2 gene located on chromosome 3q23 has been identified as a cause of BPES (Crisponi et al., 2001). BPES is inherited in an autosomal manner, but there are also sporadic cases. There is a FOXL2 mutation in 75% of individuals affected by BPES, whereas 25% of cases may have no affected family member. In a study on 33 patients with BPES, chromosomal abnormality was detected in only 24% of the cases in cytogenetic analysis. The cases with chromosomal abnormality included both sporadic and familial cases. Thus, a lack of family history does not exclude a BPES diagnosis.
In conclusion, the evidence provided in this single case report is insufficient to indicate that these findings are definitively associated with the clinical condition BPES. However, the careful recording of the phenotype in this case will provide clinicians with the opportunity to evaluate similar cases in the future and provide further evidence of whether these findings are in fact a part of BPES. In addition, we present this case because it is the first reported BPES case in Turkey.