
Abstract
Osteopetrosis (OP) is a clinically and genetically heterogeneous disorder characterized by increased bone density. Associations between OP and other clinical entities are rare but include muscular degeneration, Dandy-Walker syndrome, craniosynostosis, and poikiloderma. Infantile OP has also been diagnosed in a group of infants with neuronal storage disease. An association between OP and juvenile xanthogranuloma (JXG) has never been previously reported. Herein we present a case of an intermediate form of OP in a newborn who presented with hepatosplenomegaly and pancytopenia. Histologic evaluation of a bone marrow biopsy demonstrated abnormally thickened bony trabeculae. A liver biopsy demonstrated prominent expansion of portal areas by a histiocytic infiltrate expressing CD45, CD14, CD68, CD163, factor XIIIa, and fascin, while the biopsy was negative for S100 and CD1a. These findings were those associated with JXG. Genetic testing demonstrated a mutation involving the Pleckstrin homology domain-containing family M member 1 (PLEKHM1) gene. A different mutation in this gene has been previously reported in one other patient with OP. Our case is the 2nd reported case with PLEKHM1 mutation in a patient with a mild form of OP. It also demonstrates the 1st reported occurrence of OP concomitantly with JXG.
INTRODUCTION
Osteopetrosis (OP) is a clinically and genetically heterogeneous disorder characterized by increased bone density. This is brought about by a defect in the breakdown of extracellular matrix in the resorption lacunae of osteoclasts, resulting in a highly dense bone with the known clinical and pathological features of OP involving the hematologic, neurologic, and skeletal systems [1,2]. Three clinical forms of OP have been described: infantile malignant OP (autosomal recessive), intermediate mild OP (autosomal recessive), and an adult benign OP (autosomal dominant) [1]. Several rare associations have been reported to occur with the infantile malignant form of OP. These include the occurrence of OP in infants with a neuronal storage disease, such as Dandy-Walker syndrome, and in infants with craniosynostosis [3–5]. Migliaccio and colleagues [6] reported a case of a 10-year-old child with an intermediate form of OP and poikiloderma. On the other hand, a number of associations have been documented in the context of juvenile xanthogranuloma (JXG), including neurofibromatosis type 1 and juvenile chronic myelomonocytic leukemia [7,8]. To our knowledge, an association between JXG and OP has never been reported.
Currently known genes with mutations presenting clinically as OP include the following: TCIRG1, CLCN7, OSTM1, LRP5, TNFSF11, CAII, and NEMO genes [1,9]. Pleckstrin homology domain–containing family M member 1 (PLEKHM1) is a recently reported gene with a mutation in a patient with an intermediate mild OP [2]. For a long time, incisor-absent rats have been used as a model for human OP. The genetic defect in these rats has only recently been identified and involves the PLEKHM1 gene. This same mutation has also been demonstrated in a child with an intermediate form of OP [2].
We present a case of an intermediate form of OP with concomitant JXG in a neonate who harbored a heterozygous mutation of the PLEKHM1 gene.
CASE REPORT
This patient presented as a 1-day-old baby boy born at 38 weeks in gestation to a 27-year-old gravida 1, para 2 mother. The pregnancy was uncomplicated. Routine maternal infectious serologies were negative. Delivery was complicated by prolonged labor, moderate meconium-stained amniotic fluid, prolonged rupture of membranes, and fetal cardiac decelerations. Delivery was accomplished via urgent primary C-section. Apgar scores were 7 at 1 minute and 8 at 5 minutes of life. Shortly after birth, the baby was noted to be pale and in moderate respiratory distress, requiring nasal continuous positive airway pressure for support. A complete blood count obtained at a few hours of life was significant for severe anemia and thrombocytopenia (ie, hemoglobin of 6.9 g/dL, hematocrit of 22%, and a platelet count of 10 000). A hematology consultation was obtained.
The physical examination was remarkable for hepatosplenomegaly, bruising on the scalp, and a petechial rash. The infant required transfusions of red blood cells and platelets on the 1st day of life and subsequently required serial transfusions to sustain adequate hemoglobin and platelet levels. His peripheral blood smear showed poikilocytosis with no blasts and normal-appearing platelet morphology. Blood culture, TORCH (toxoplasmosis, other infections, rubella, cytomegalovirus, herpes simplex virus) titers, Parvovirus, and human immunodeficiency virus studies were all negative. An echocardiogram on day 1 of life demonstrated normal cardiac structure and function. Abdominal ultrasound confirmed hepatosplenomegaly but was otherwise normal. Head computed tomography (CT) on the same day showed no intracranial hemorrhage. On day 9 of life, a technically difficult tibial bone marrow biopsy was performed to evaluate the anemia and thrombocytopenia. The biopsy showed much crush artifact, which rendered evaluation difficult, but it also showed better preserved areas demonstrating thickened bone trabeculae (Fig. 1A). No definite evidence of JXG was seen. The aspirate smears showed predominantly normoblastic maturation with an increased myeloid:erythroid ratio.
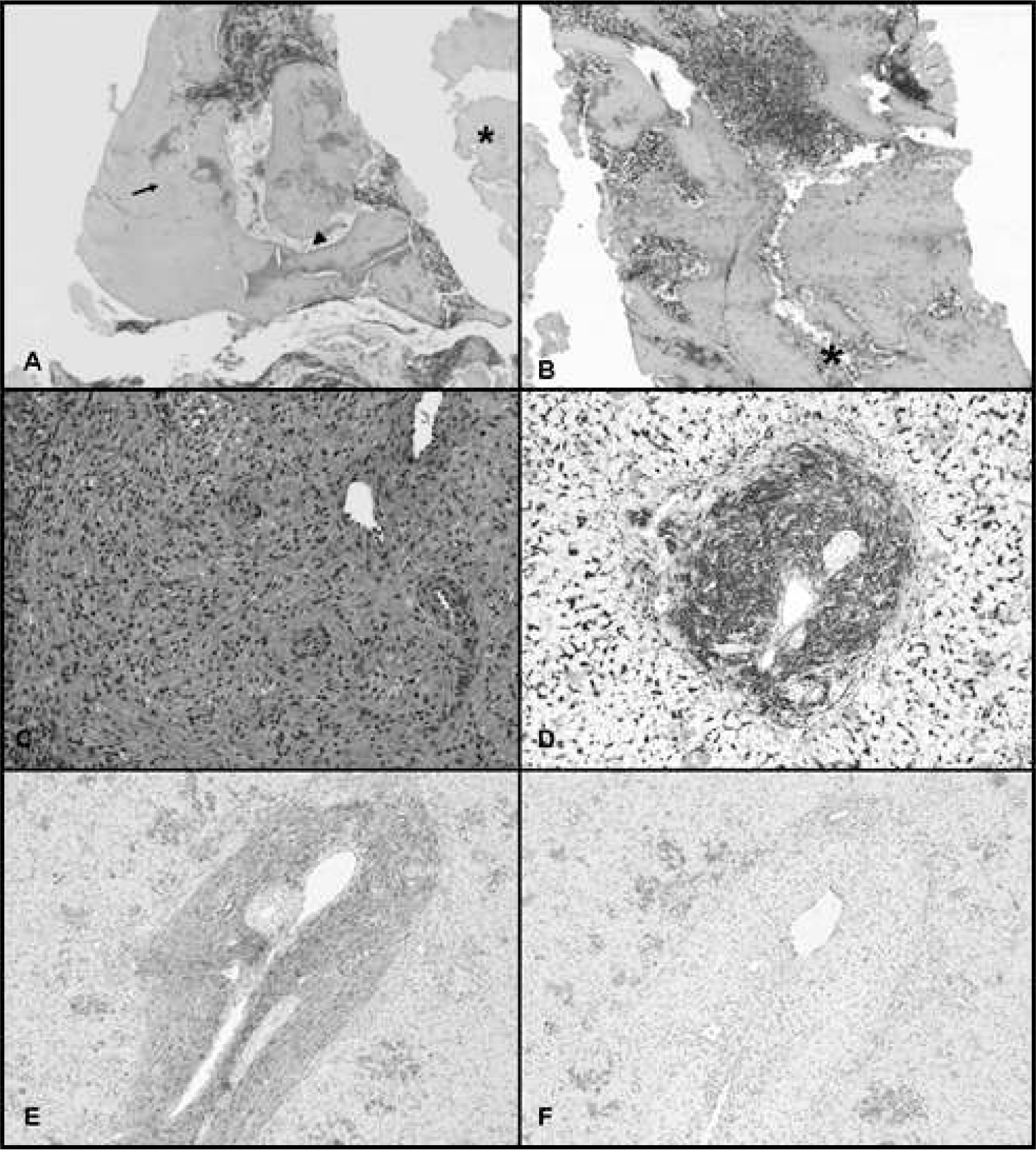
A repeat bone marrow biopsy from the iliac crest on day 11 of life showed thickened bony trabeculae, consistent with a diagnosis of OP (Fig. 1B). It highlighted a very dense, thickened bone and bony trabeculae with some crushed bone and variable marrow space ranging from almost no marrow to minimal marrow to focally marrow areas with hypercellular (>80%–90%)-appearing trilineage hematopoiesis. No evidence of malignancy, storage disease, or infection was seen. Even in areas of normal trilineage hematopoiesis, the surrounding bony trabeculae showed markedly abnormal thickening and overgrowth. Osteoclasts were noted as well as Howship's lacunae. There was no definite change in the number of osteoclasts present. There was no obvious fibrotic effacement of medullary spaces, possibly related to the very young age of this patient.
X-rays of upper extremities on day 7 of life were normal, with no evidence of generalized bone sclerosis, modeling deformity, widened or irregular metaphyses, or fractures. Follow-up head CT scan also showed normal bone density. An orbital CT scan was normal, with no optic canal narrowing. Magnetic resonance imaging of the face and orbits also showed no narrowing of the optic foramina. On day 11 of life, a liver biopsy was obtained as a result of the unclear etiology of the infant's hepatosplenomegaly. It demonstrated extensive and multifocal extramedullary hematopoiesis with clusters of interspersed myeloid precursors. There was also mild to moderate intracanalicular cholestasis as well as focal microvesicular steatosis. Most significantly, there was a prominent expansion of the portal areas by a florid proliferation of histiocytes with interspersed eosinophils and lymphocytes (Fig. 1C–F). The histiocytes were characterized by bland oval nuclei and abundant foamy eosinophilic cytoplasm. The bile ducts were unremarkable and devoid of infiltration by the proliferating histiocytes. The histiocytes were highlighted by immunohistochemical staining with CD45, CD14, CD68, CD163, factor XIIIa, and fascin. No staining for S100 or CD1a was identified. The findings in the liver were those of JXG.
The patient was later found to be heterozygous for a point mutation in the PLEKHM1 gene, exon 7. Mutations involving this gene in the context of OP have been previously associated [2,10] with 2 other cases of intermediate OP. The patient's mother was also heterozygous for the same mutation; however, she was phenotypically normal. No mutation was demonstrated in the father.
The patient was discharged from the hospital at approximately 6 weeks of life. His hemoglobin and platelet counts stabilized, and he required no further transfusions. He was subsequently seen in follow up on a monthly basis, with no major interventions necessary. Currently he continues to be hematologically stable and developmentally appropriate at 8 months of age.
DISCUSSION
Osteopetrosis represents a rare genetic defect. Although recognized for a long time, its pathogenesis has only recently been explored. We have come to realize that this is a clinically and genetically heterogeneous disorder. A defect in resorption of extracellular matrix within the resorption lacunae of osteoclasts results in highly dense bone with the known clinical and pathological consequences of OP, including hematologic, skeletal, and neurologic manifestations. Osteopetrosis is classified according to the severity of disease, age of onset, and pattern of inheritance [1,2]. Three clinical forms have been described [1]: infantile malignant OP (autosomal recessive), intermediate mild OP (autosomal recessive), and adult benign OP (autosomal dominant).
Infantile malignant OP is the least common form, with an incidence of 1:200 000 to 1:300 000. It presents in the 1st year of life with hematopoietic deficiencies, bony abnormalities, and neurological manifestations [1]. The bone is thickened but brittle and can be detected by radiologic imaging showing metaphyseal widening of long bones. Children succumb to their disease by the age of 4 years if left untreated [1]. Bone marrow transplantation is aimed at replacing the patient's osteoclasts with functioning donor osteoclasts, thus offering a potential cure of the underlining genetic defect [11]. Therapeutic attempts utilizing Calcitriol and interferon gamma as an alternative to bone marrow transplantation were of limited benefit [11]. Mutations documented in the infantile malignant form of OP include mutations in TCIRGI, CLCN7, and OSTM1 genes [1]. The most common mutation involves the TCIRGI gene that encodes the α-3 subunit of adenosine triphosphatase, which involves proton release to the resorption lacunae of osteoclasts. The protons released are transported to the resorption lacunae through a calcium channel located in the osteoclast ruffled border membrane. This chloride channel is encoded by the gene CLCN7, another gene reported to be mutated in patients with infantile malignant OP. The OSTM1 gene has a possible role in osteoclast cytoskeletal reorganization of the ruffled border [1].
In intermediate mild OP, patients generally present later in life, with milder phenotypic expression and better survival rates. The known genes involved in the intermediate form of OP are the CLCN7 and PLECKHM1 genes. Patients with intermediate OP and CLCN7 gene mutation have a partial loss of function of the gene, with resultant decreased chloride channel conductance [1]. In addition to our case involving PLEKHM1 gene mutation, there is one other reported case [2], although this other case involves a different mutation than the one reported in our case.
Autosomal dominant adult OP has a benign clinical course. Cases are detected incidentally during radiologic evaluation for other reasons. Some of the documented mutations in these patients involve the CLCN7 and LRP5 (a low-density lipoprotein receptor-related protein 5) genes [1]. Yet other forms of OP include a severe osteoclast-poor form, a transient infantile form, OP with renal tubular acidosis (carbonic anhydrase II mutation), OP with neuronal anomalies, OP with anhydrotic ectodermal dysplasia, and OP with Glanzmann's thrombasthenia [1]. Our case represents the 1st described case of OP with JXG.
Our understanding of the genetic and pathophysiological changes in OP is only in its infancy. To date, only a handful of mutations have been documented in OP, including those involving the following genes: TCIRG1, CLCN7, OSTM1, LRP5, TNFSF11, CAII, and NEMO [3,9]. An animal model of OP involving incisor-absent rats has been used for some time to study the pathophysiologic changes in OP. These rats seem to have an intermediate form of OP, with a partial resolution of symptoms that has been documented between 30 and 50 days of life [2].
The PLEKHM1 gene has only recently been identified as the defective gene in those rats showing a homozygous deletion of a cytosine in the 4th coding exon. This deletion causes a frameshift mutation, resulting in a truncated protein. PLEKHM1 gene has been mapped to 17q21.3 in humans [12]. The gene product has a role in late endosomal/lysosomal vesicles, as demonstrated by Van Wesenbeeck and colleagues [2]. This might have a role in vesicular transport of osteoclasts, as suggested by these authors [2].
Van Wesenbeeck and colleagues [2] reported the 1st human case with OP, in which the underlining genetic defect was mapped to the PLEKHM1 gene. Their case had a homozygous G→A transition at position +1 of the donor splice site of the PLEKHM1 gene intron 3. At 7 years of age, this female patient was noted to have cortical sclerosis of the pelvic bones, vertebral endplates, and the meta-diaphyses of the distal femora, tibiae and fibulae, and the proximal fibulae and tibiae. Broadening of the involved long bone segments was noted. Both of the patient's parents were carriers of the mutation and showed no evidence of disease. She had 2 siblings, an older brother who was heterozygous for the mutation and phenotypically normal and an asymptomatic brother who was homozygous for the mutation. A PLEKHM1 gene with loss of function has been suggested to play a role in vesicular transport within osteoclasts. Del Fattore and colleagues [10] reported on another patient with a PLEKHM1 gene mutation, a 39-year-old female with a long history of skeletal abnormalities. She initially presented at the age of 3 years and was diagnosed with rickets. At the age of 28, she was given a diagnosis of OP with vitamin D3 deficiency. At the age of 39, she was found to have generalized osteopenia and localized skull sclerosis. She harbored a heterozygous C to T change at position 2140 of the PLEKHM1 gene.
As noted, our patient is heterozygous for a point mutation in exon 7 of the PLEKHM1 gene. His phenotypically normal mother was also heterozygous for this exon 7 mutation. The father tested negative for known mutations implicated in OP. Our case demonstrates a defect in bone homeostasis that can be explained by the harbored mutation in PLEKHM1. The exact role this mutation plays in the pathogenesis of the bone changes, in contrast to the one reported by Van Wesenbeeck and colleagues [2], is not known. Our case, like that described by Van Wesenbeeck and colleagues, involves a milder form of clinical presentation of OP. Although the case reported by Del Fattore and colleagues [10] does not represent an example of OP, it highlights the fact that abnormalities of the PLEKHM1 gene have consequences with regard to the normal development of bone.
The histologic evaluation of bone in patients with OP demonstrates thickened bone trabeculae with central calcifying cartilage. Osteoclast number is not affected since the disorder involves abnormal function rather than abnormal quantity [13]. Reports have indicated that a paucity of osteoclasts may be seen; however, many cases show an abundance of osteoclasts. There is a failure to resorb calcified cartilaginous foci, and such foci may show a deficiency of osteoclasts [14].
The pathologic differential diagnosis of OP includes pycnodysostosis, which is a rare disorder of osteoclasts in which patients present with generalized osteosclerosis, and this disorder can mimic OP. It is caused by a loss of function mutation in the cathepsin K gene that encodes a proteinase involved in degradation of bone protein, such as collagen type I [1]. It is associated with an abnormal skull, with open sutures and bone fragility. The clinical presentation as well as the underlying genetic abnormality help differentiate it from OP [15].
Juvenile xanthogranuloma is a dendritic cell-related proliferation with localized or generalized lesions. In Dehner's [7] series of 174 cases of JXG, two thirds of the cases presented with a solitary skin lesion, with 42% of those lesions presenting in the head and neck region. The lesion is usually a <1-cm nodule or plaque with flesh-colored, erythematous, or yellow appearance [7]. A solitary extracutaneous lesion was only seen in 5% (9 of 174 cases) of the cases. In those cases, 7 lesions were in the head and neck region, 1 was in a mainstem bronchus, and 1 was in a lumbar vertebra [7]. Liver involvement in JXG can be an isolated finding or part of a systemic disorder; however, hepatic involvement usually occurs in the systemic form of JXG [8].
Juvenile xanthogranuloma is composed of an infiltrate characterized by a mixture of mononuclear cells, spindle cells, and multinucleated giant cells with or without Touton giant cells. The proportion of the different cell components varies between cases. The extracutaneous lesions are composed mainly of mononuclear cells, a mixture of mononuclear and spindle cells, or pure spindle cells. They are noted to have rare or no giant cells, in contrast to cutaneous lesions. Mononuclear cells are characterized by round to reniform nuclei with uniform chromatin pattern [7]. Cytoplasmic vacuoles within mononuclear and giant cells imparting the xanthomatous appearance of these lesions can be seen but are less frequent than the nonvacuolated forms [7]. A background of inflammatory cells can be seen, in particular eosinophils. Atypia and mitoses are reported in 10% of cutaneous lesions in the Dehner series [7]. In the liver, JXG is characterized by a predominantly portal expansion. A giant cell hepatitis can be seen in these cases [7,8]. Immunohistochemical evaluation of these lesions shows positivity for CD68, CD168, factor XIIIA, fascin, CD14, and vimentin The cells are negative for Sl00 and CD1a, although weak staining with S100 can be noted focally in some cases [7,8].
The cells of JXG are thought to be of dendritic cell origin. However, evidence shows that Kupffer cells, a macrophage derivative, and dendritic cells can interchange. Both macrophages and dendritic cells are derived from the same stem cells. They differentiate into their mature forms depending on the cytokine environment to which they are exposed. Interdifferentiation from macrophage to a dendritic cell, and vice versa, has been shown [8,16].
In addition to JXG, the differential diagnosis of a histiocytic infiltrate in the liver includes Langerhans cell histiocytosis, infections, metabolic, or storage disease. Langerhans cell histiocytosis is characterized by a striking biliary epithelial tropism, a feature not seen in JXG. The infiltrating cells stain with S100 and CD1a [8]. Atypical mycobacterial infections can mimic a neoplastic histiocytic hepatic infiltrate. An infectious etiology can be ruled out by taking into account the clinical presentation of the patient as well as through the use of negative special microbial stains and serological tests. A storage disorder with a histiocytic infiltrate of the liver can be ruled out by identification of storage material by light microscopy, electron microscopy, and biochemical tests and through genetic evaluation of the suspected disorder.
There are limited data on the outcomes in patients with solitary hepatic JXG. Jaffe [8] encountered 3 cases of hepatic JXG; 2 patients were alive without the disease following treatment. One of the patients was treated with cyclosporin and the other with etoposide/prednisone. The 3rd patient was lost to follow up.
Association with JXG has been consistently reported in the literature [7,8] to occur with neurofibromatosis type 1 and juvenile chronic myelomonocytic leukemia. Infantile OP has been reported in a group of infants with neuronal storage disease. The storage material was found to be composed of lipids, oligosaccharides, and polylactosamines [17]. These disorders have been shown to harbor CLCN7 or OSTM1 mutations. The coded molecules of these genes seem to be related in terms of function. Ostm1 facilitate the localization of CLC7 to the lysosome and provide stability to the molecule [17]. Rare associations with muscular degeneration, Dandy-Walker syndrome, craniosynostosis, and poikiloderma need further clarification [1]. This is the 1st reported case of JXG associated with OP. The possibility that the genetic defect seen in our case of intermediate OP is related to the hepatic histiocytic infiltrate is raised. Recent data demonstrating an interchange between dendritic cells, the cell of origin of JXG, and macrophages, an osteoclast precursor, may explain the findings in this patient with PLEKHM1 gene mutation [8,16,18]. A definite attribution of JXG to the genetic disorder causing the OP in our case is not possible. One is always faced with the possibility of coincidental association rather than a common etiological process. In conclusion, we report, to our knowledge, the 1st association between OP and JXG in a neonate. Our case is the 2nd case involving PLEKHM1 gene mutation as the defect in a case of intermediate OP.
Footnotes
ACKNOWLEDGMENT
The authors would like to thank Dr Ronald Jaffe for his review of the liver biopsy in consultation.