
Abstract
The development of an acardiac twin in a monochorionic multiple pregnancy is a rare and severe complication of abnormal placental vascular anastomoses. These malformed fetuses present with a very bizarre morphology and a plethora of different malformations. However, all acardiac twins show either a complete absence or an anlage of the heart. Cerebral development is usually poor. We report, according to our review of the literature, for the first time, a very unusual case of acardius with features of acardius amorphus and acormus (fused head and malformed axial skeleton without macroscopically detectable internal organs) with lobar holoprosencephaly and intracerebral pigmented retina-like tissue.
Keywords
BACKGROUND
The development of an acardiac twin, that is, the twin reversed arterial perfusion (TRAP), one of the most severe human malformations, occurs in 1 case in 30 000 pregnancies [1]. In addition to a bizarre morphology and serious parenchymal malformations, complete absence or only an anlage of the heart is the main characteristic feature of acardiac twins. Hence, the acardiac twin depends on blood transfusion from the structurally normal co-twin (“pump twin”). Through a superficial artery-toartery network, seldom an artery-to-vein placental anastomoses, deoxygenated arterial blood runs retrograde from the pump twin into the arterial system of the other twin (“recipient twin,” ie, the acardiac twin). From there, blood flows through the umbilical cord and venous-venous anastomoses back to the co-twin (pump twin), whose heart pumps for both twins (Fig. 1).
Based on the morphology of the acardiac twin, four types are to be distinguished [2]. Acardius anceps (8%) displays a fairly well-developed body and extremities but only a rudimentary head with an imperfect face. Of the internal organs, the genitourinary system especially can be highly differentiated. It is postulated that the other types begin as acardius anceps but, due to poor oxygen and nutrient supply, evolve into less well-differentiated types [3]. Acardius acephalus, representing the most common form (62%), is characterized by the lack of a head, thorax and thoracic organs, and often upper limbs but the presence of a well-developed pelvis and lower limbs. Acardius acormus, the rarest form (5%), presents with a fetal head and sometimes a malformed axial skeleton. Internal organs are usually missing. Acardius amorphus (25%), the least developed type, has no recognizable human form and appears as a skin-covered, shapeless, often edematous mass without recognizable organs. Sometimes, some form of axial structures can be detected, while regularly shaped bones are absent [4].
We report an acardiac fetus with features of acardius acormus and acardius amorphous showing lobar holoprosencephaly with a relatively well-developed brain and with intracerebral retina-like pigmented tissue, some residual pharyngeal structures, and axial skeletal bones without limbs. The monochorial placenta was diamniotic, measured 17 ×14.45 cm, and weighed 480 g (normal weight for 31 weeks of gestation: 195–400 g). Both of the twins’ umbilical cords inserted marginally and less than 0.5 cm from each other, and each cord had three vessels. No abnormalities were detected, either macroscopically or microscopically.
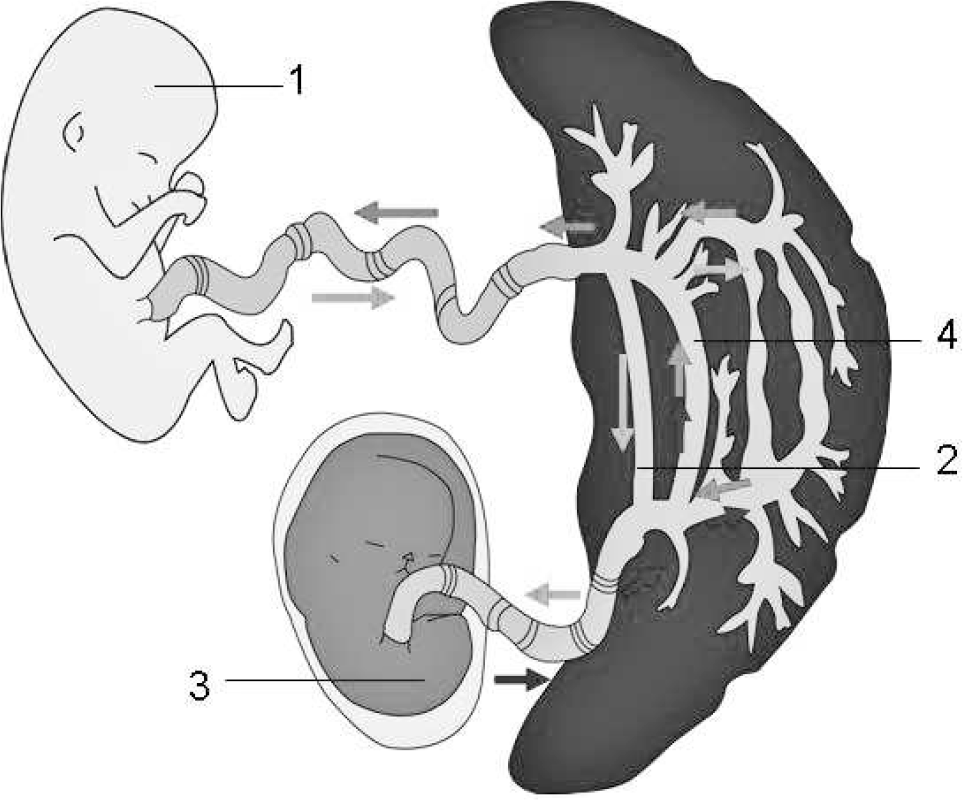
Blood flow is shown in an acardiac twin. Retrograde arterial blood from the pump twin (1) flows through an anastomosis (2) toward the acardiac twin (3). From there, it flows through the umbilical cord and venous-venous anastomoses back to the co-twin, whose heart pumps for both twins (“pump twin”).
CASE HISTORY
Clinical presentation
A 33-year-old woman presented to Bonn University Hospital at 12 weeks and 6 days of gestation. Ultrasonography revealed a morphologically regularly formed fetus and a dysmorphic twin with rudimentary cardiac structures and a developed head with a malformed brain. The diagnosis of TRAP fetus was established. The pumping fetus was followed closely with serial ultrasonography, Doppler sonography, and echocardiography (Fig. 2). After 31 weeks, following a spontaneous premature rupture of membranes, a healthy female neonate (weight, 1490 g; length, 43 cm; head circumference, 30 cm) was delivered by cesarean section, as was a skin-covered ovoid, discus-like mass, which weighed 902 g and measured 19.0 ×13.5 ×8 cm.
Pathomorphological findings
Autopsy of this ovoid mass revealed a cephalic structure with two eyelids and a proboscis and a stoma at the upper end (Fig. 3). Radiography and computed tomography (CT) scans (Fig. 2) showed a well-developed cranium with nasal and mandibular bones and a skeleton consisting of a ribcage with flattened ribs and no sternum, two abnormally formed scapulae, multiple vertebrae, and a right iliac bone. Bones of upper or lower limbs could not be identified. A massive dorsal cystic lesion in the upper region, resembling a cystic hygroma, surrounding the “cephalic” structures was also identified. Eyeballs and orbital structures were absent. In addition, in the upper part of the body, an oral cavity with a tongue and transition to a pharyngeal region with laryngeal structures and a structure that macro- and microscopically resembled an esophagus could be distinguished. In addition, a trachea with a rudimentary bronchial bifurcation could be identified, although normal bronchi were absent. Furthermore, a large pancreas and massively dilated intestinal loops were present. Two umbilical arteries joined to form a rudimentary aortic arch, with multiple small vessels reaching the head region. Stomach, liver, kidney, spleen, or lung could not be detected, either macroscopically or microscopically.
Unexpectedly, a specimen taken from the “abdominal” center of the acardius, that is, in the neighborhood of the gut, displayed histological evidence of a rudimentary heart (Fig. 4). Additional microscopic specimens from this area showed an adrenal gland, an ovary, and rudimentary plump ovarian fimbriae as well as several paraganglia.
Neuropathological findings
In the cranial cavity, the anterior, medial, and posterior fossae were present, but crista galli, cribriform plate, and optic canals could not be identified. A rudimentary falx cerebri could be detected.
The formalin-fixed brain weighed 180 g (corresponding to standard values of the 28th to 29th week). External macroscopic examination revealed no clear malformation of cerebellum, brain stem, and spinal cord. In the interhemispheric fissure, the corpus callosum could not be identified (Fig. 5). The cerebral hemispheres over the convexities showed an advanced gyrification without evidence of malformations, necroses, or hemorrhages. A basal view of the brain showed the absence of olfactory and optic nerves.
Only the most rostral basal parts of the frontal lobes were partially fused. Here, the tissue appeared firm and more whitish in color (Fig. 5). Due to poor preservation, the brain tissue was extremely fragile, and the sectioning was difficult. Frontal sections of the brain revealed a marked dilatation of the ventricular system and confirmed fusion of the infolded most rostral basal midline structures (Fig. 6). There was no clear identification of basal ganglia and thalami. Agenesis of corpus callosum was confirmed. Necroses and hemorrhages could not be identified. Sections of the infratentorial segment showed no abnormalities. In the midbrain, the aqueduct could be identified and was patent.
Microscopically, the cerebral cortex showed a focal polymicrogyria-like architecture (Fig. 7A–D). Furthermore, multiple nodules of immature neuroepithelial cells were found throughout the cerebral layers (Fig. 7A, inset). Foci of leptomeningeal glioneuronal heterotopia were present in several brain regions, including the parietooccipital lobe (Fig. 7B,C) and temporal cortex, the brain stem, and the spinal cord. Foci of proliferating reactive meningothelial cells were also identified, mainly in malformed basal midline areas. No histological anomalies were found in the cerebellum.
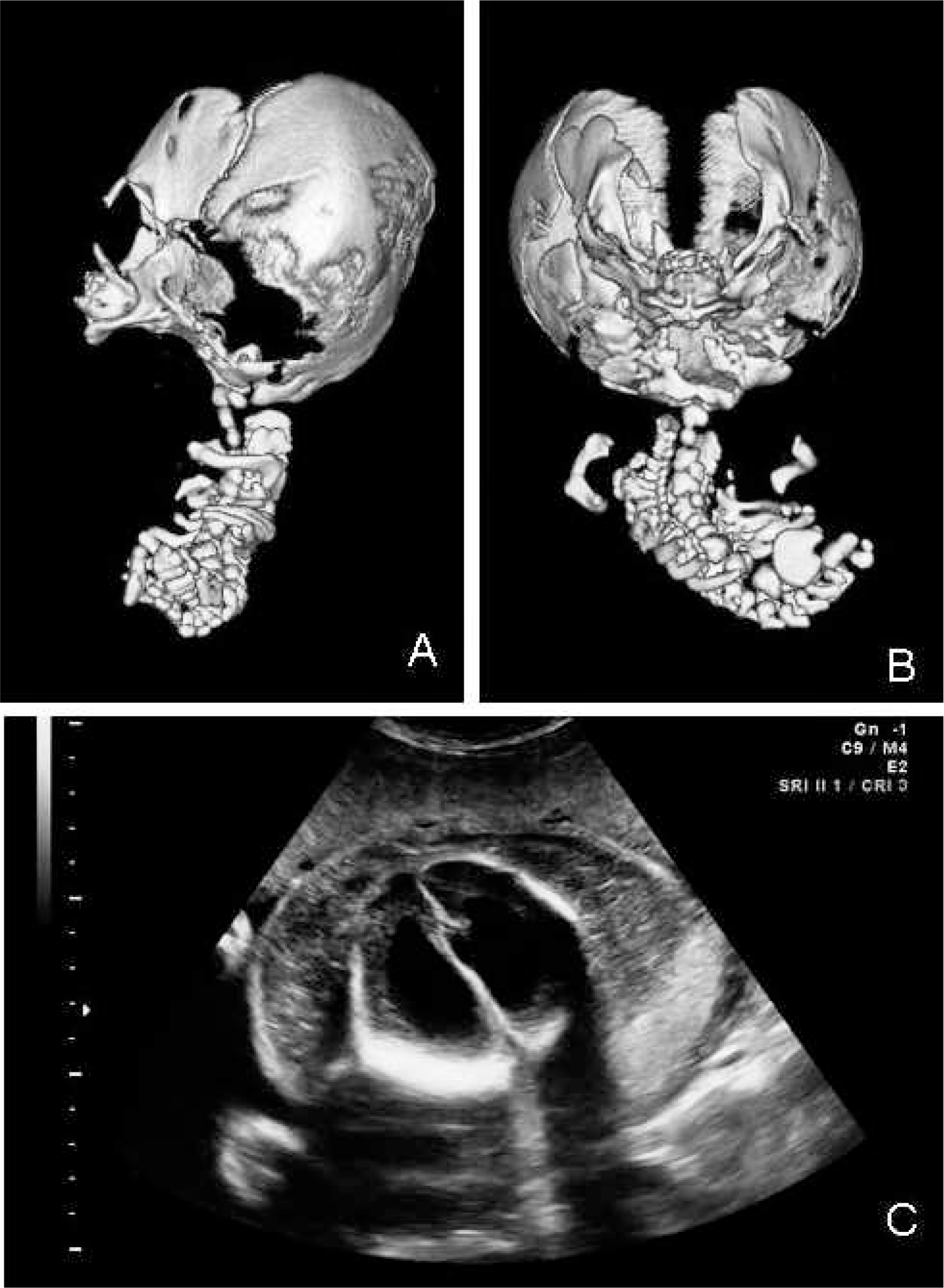
Computed tomographic (CT) scan and ultrasonography studies are shown. Lateral and axial CT reconstructions (
The infolded and fused rostral basal region of the brain was composed of areas of immature neuroepithelial cells partially surrounded by cortical nervous tissue and separated by fibrous septa (Fig. 6B). In this area, we also identified intermingled bands and cords of pigmented cells without a definite spatial orientation (Fig. 6B, inset) as well as fibrous and epithelial tissue, probably from skull base structures. The findings corresponded to a lobar holoprosencephaly with fusion of only the most rostral frontobasal areas, without clear identification of basal ganglia and thalami, with agenesis of corpus callosum and olfactory aplasia and anophthalmia.
DISCUSSION
Acardiac twin syndrome affects monozygotic twins, where due to the absence of a functioning heart, one twin fails to develop normally.
The first cases were reported in the 16th century, and to our knowledge, no more than 500 cases have yet been described. The frequency of an acardiac twin is estimated at 1 case in 30 000 deliveries and 1% of monochorionic twins [3,5]. Besides the classification described above, a hemiacardius heart (incompletely formed heart) and a holoacardius (absent heart) can be distinguished.
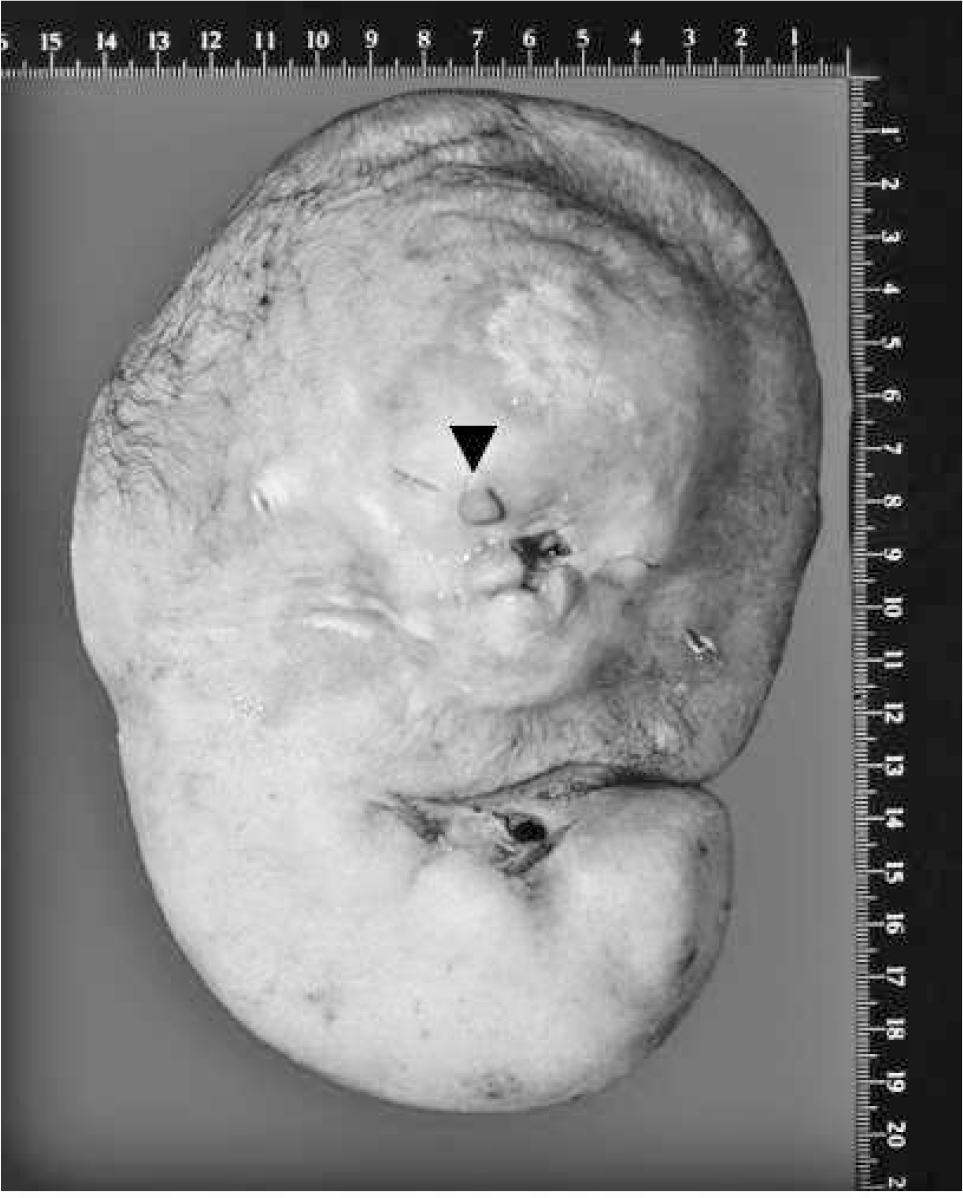
Acardius is shown, including a mainly discus-like shape with a proboscis (arrow), eye lids, and a stoma.
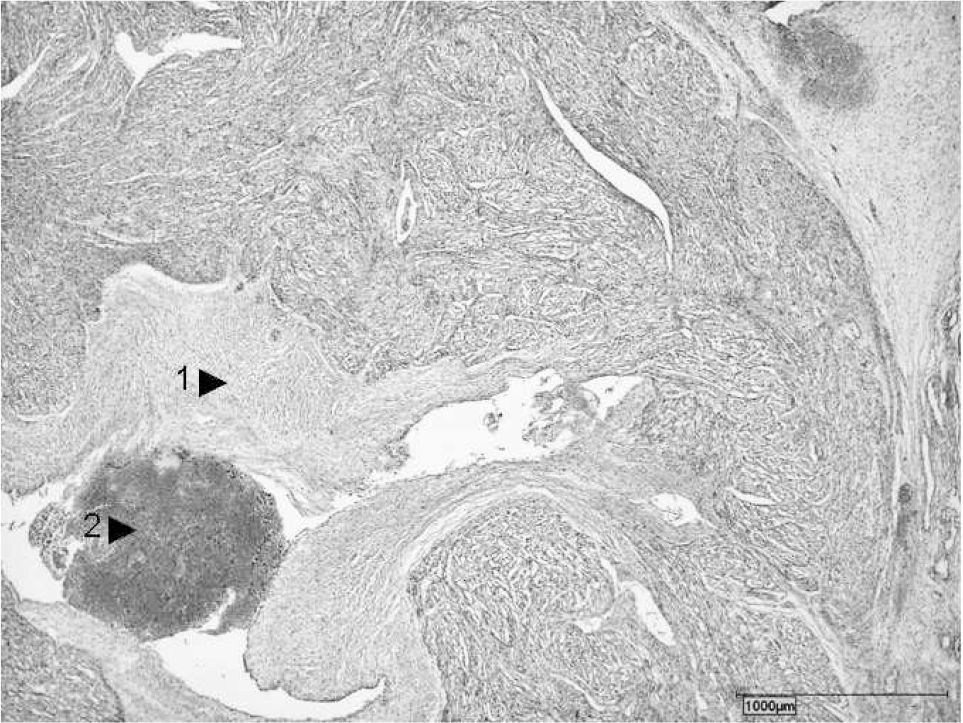
Histological features of the heart include rudimentary anlage of the heart with endocardial fibrosis (1) and thrombus (2) (hematoxylin-eosin stain).
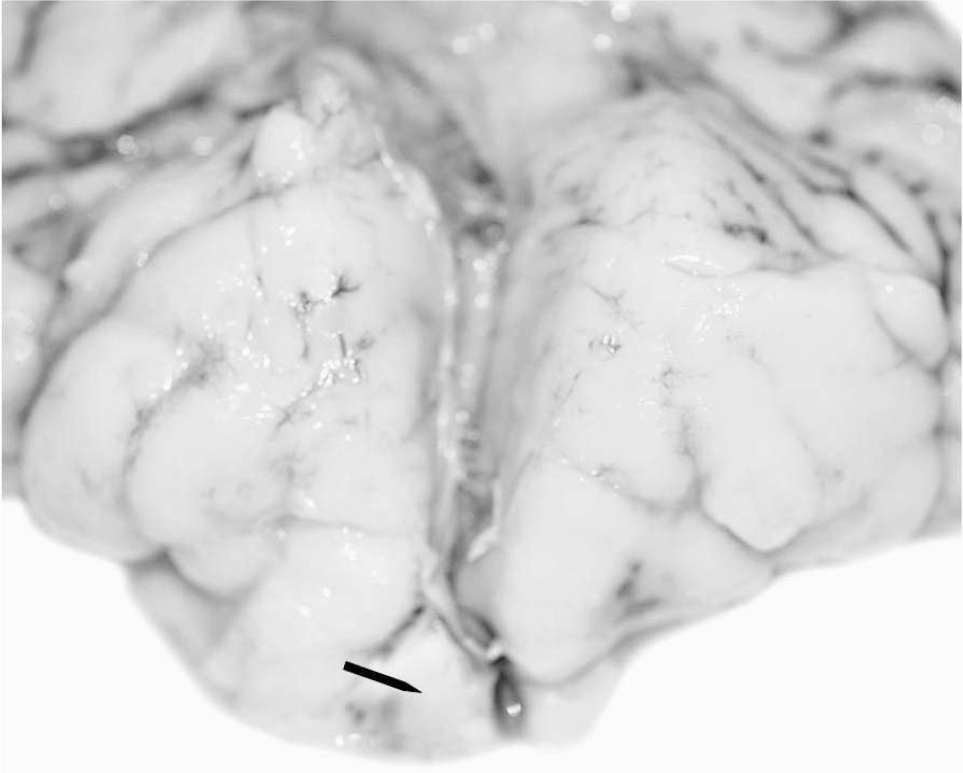
Frontopolar view of the brain shows advanced gyration, lack of corpus callosum, and fusion of the rostral part of the frontal lobes by a firm and more whitish tissue (arrow).
According to our review of the literature, the phenotype of the acardius twin presented here has not yet been described because (1) it displays features of both acardius amorphus (shapeless mass without macroscopically recognizable organs) and acardius acormus (radiologically well-developed cranium and mouth-like structures as well as eye lids and malformed axial skeleton), (2) it presents an unusually well-developed brain with lobar holoprosencephaly and with intracerebral retina-like pigmented tissue. The first point raises the question of whether the so-called Latin classification is still useful. Considering the usual lack of differentiated internal organs in acardius acormus and acardius amorphus, the presence of a relatively well-developed brain with holoprosencephaly and intracerebral retina-like pigmented tissue is unique. Only a few reports describe cerebral structures in acardiac fetuses (Table 1). The incomplete development of the brain is considered a result of inadequate blood perfusion, which is unable to sustain complete central nervous system development [1,6–11]. This theory is called into question by our findings of a considerably well-developed brain. While the infratentorial structures (including cerebellum and spinal cord) are often present and may show a broad spectrum of malformations, the forebrain is often absent or sometimes greatly malformed (eg, holoprosencephalic features and absence of the midline structures) [10]. Cases with partial or complete development of forebrain structures and cerebral cortex seem to be exceptionally rare. Petersen and colleagues [11] reported a case of an acardiac fetus with a nearly regular external appearance, which in our opinion, therefore should be classified as acardius anceps and not acardius acormus, with a full cortical development with only slight architectural anomalies including polymicrogyria and ectopic mesoglial nodules. These types of cerebral malformations are hypothesized to be related mainly to the disruption of the glia-pia boundary and to disturbance of the migration process of the immature neuroepithelium [12]. As described in other cases [9], unfolding and migration disturbances may coexist. Our case showed features of lobar holoprosencephaly with olfactory aplasia and the presence of an interhemispheric fissure separating relatively well-formed hemispheres, with only a small focal infolding and fusion of the most rostral frontobasal region. The latter finding is still compatible with the diagnosis of lobar holoprosencephaly and does not require classification as semilobar holoprosencephaly [13]. Histologically, alterations of cortical architecture with polymicrogyria-like features and leptomeningeal glioneuronal heterotopias were present.
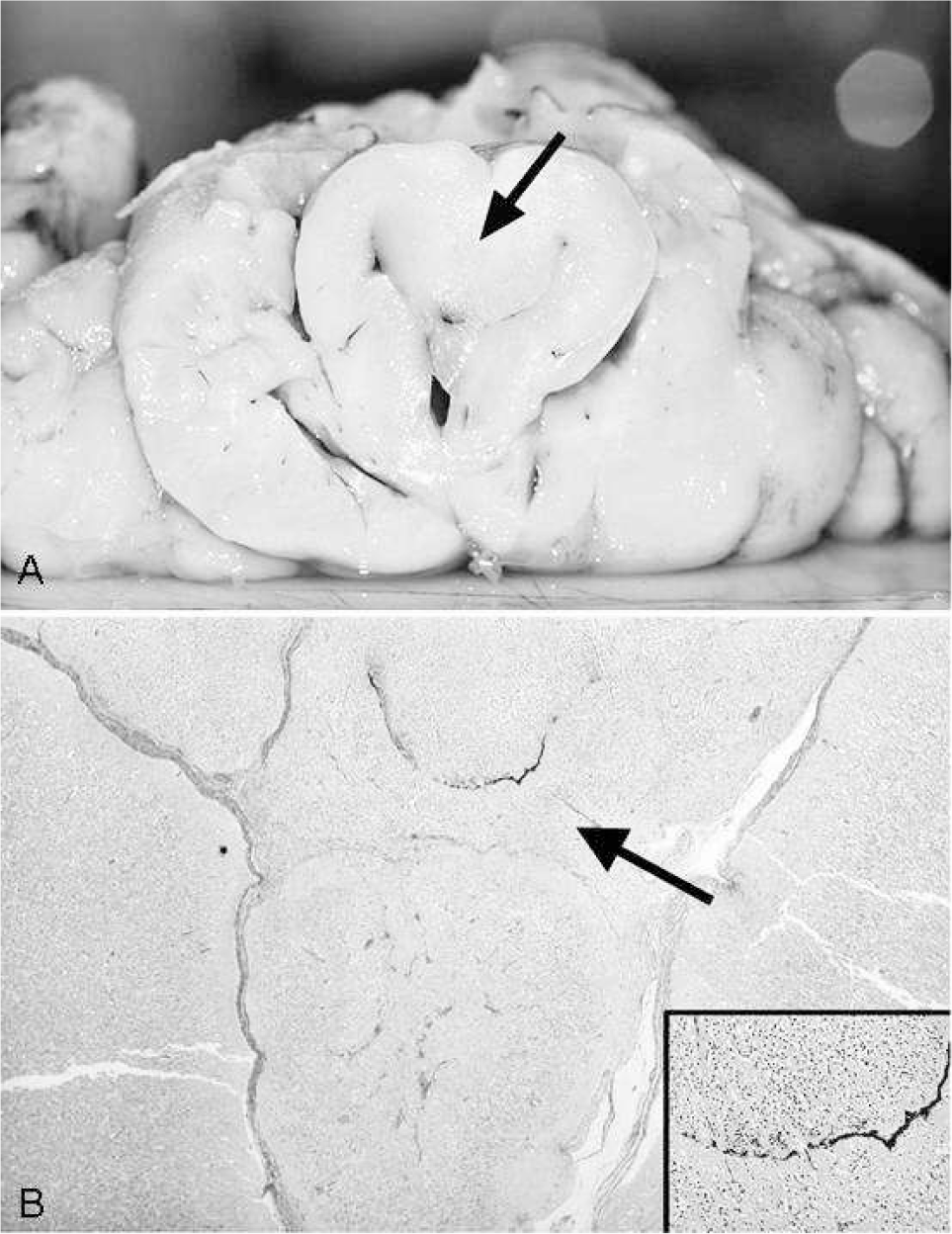
Macroscopic and microscopic features of frontobasal midline structures are shown. (
The complete absence of the intracranial optic pathway and intraorbital ocular structures (including the eye) is a common finding in acardiac fetuses and in holoprosencephaly. An incomplete development or severe malformations (including cyclopia) of ocular and orbital structures have also occasionally been reported in acardiac fetuses [6]. Anophthalmia and cyclopia are often found in holoprosencephaly [14]. In holoprosencephaly, developmental defects of facial bones including bones of the skull base can occur. Such cases may show a proboscis as in the present case [15].
In our case, intracranially, we could not identify any anatomic structures corresponding to optic pathways. However, we found in the frontobasal malformed brain areas multiple nests and cords of pigmented cells. We interpret these cells as nests of pigmented retinal tissue probably resulting from an early failure of migration processes that lead to the formation of the retina and the optic nerves. Failure of formation of orbital structures and other ocular structures also occurred. To our knowledge, intracerebral retina-like pigmented tissue, as seen in the present case, has not yet been reported in holoprosencephaly or in acardiac fetuses.
To explain the etiology and pathogenesis of the TRAP sequence, two major theories have been proposed by Benirschke and des Roches Harper [12]. One theory postulates that two separate conditions are required: (a) a severe genetic or other primary defect sufficient to cause failure of the cardiac development in 1 of the twins and (b) anastomoses formed between the vessels of the 2 umbilical cords. The second theory is based on the development of artery-to-artery anastomoses between the twins in the presence of a fused placenta early in the 1st trimester, leading to the vascular disruption of 1 twin and the development of TRAP sequence.
The most striking pathoanatomical correlate for the latter theory is the formation of superficially located vascular anastomoses (arterial-arterial and veno-venous) on the placental surface. It is a prerequisite that the pump twin's blood perfuses the placenta to become oxygenated. The oxygenated blood is pumped via (arterial-arterial) anastomoses, via umbilical arteries into the body of the acardius, ie, the blood flow is reversed such that the acardiac fetus gains its oxygen-enriched blood via its umbilical arteries. Deoxygenated blood from the acardius flows via venous-venous anastomoses into the umbilical vein of the twin fetus, merging there with oxygenated blood. Hence, the blood pumped by the pump twin's heart falls short of the usual oxygen saturation levels. Therefore, both twins suffer from deoxygenated blood [16,17].
Formation of acardius is observed more often in a monoamniotic rather than a diamniotic placenta. Usually, in the monoamniotic placenta, the distance between the insertions of both umbilical cords is smaller. Such a small distance between both umbilical cord insertions can be bridged by short anastomoses. Resistance is related linearly to the length of the vessel. The shorter the vessel, the lower the resistance [18]. Some authors claim that the distance between both insertion points helps to predict the type of acardius. A distance of approximately 1.5 cm would result in well-differentiated acardius anceps; a distance of approximately 0.5 cm would promote development of a poorly differentiated acardius amorphus [4]. As in our case, the two umbilical cords inserted in immediate proximity (<0.5 cm), and the placenta was diamniotic; the morphology of our case calls into question this resistance theory.
In contrast to the relatively well-developed brain with a minor form of holoprosencephaly, we found only very poorly developed parenchymal organs. This is astounding, as neuronal tissue reacts very sensitively to a lack of oxygen. Hence, mechanisms other than high levels of deoxygenated blood in acardius might play a crucial role in determining whether brain-like structures differentiate during pregnancy. Recently, Martinez-Frias [19] proposed that epigenetic processes involved in the early cleavage of the embryo and in blastocyst formation during early stages of development might play a crucial role in TRAP [19]. This may well be one mechanism that could explain our findings.
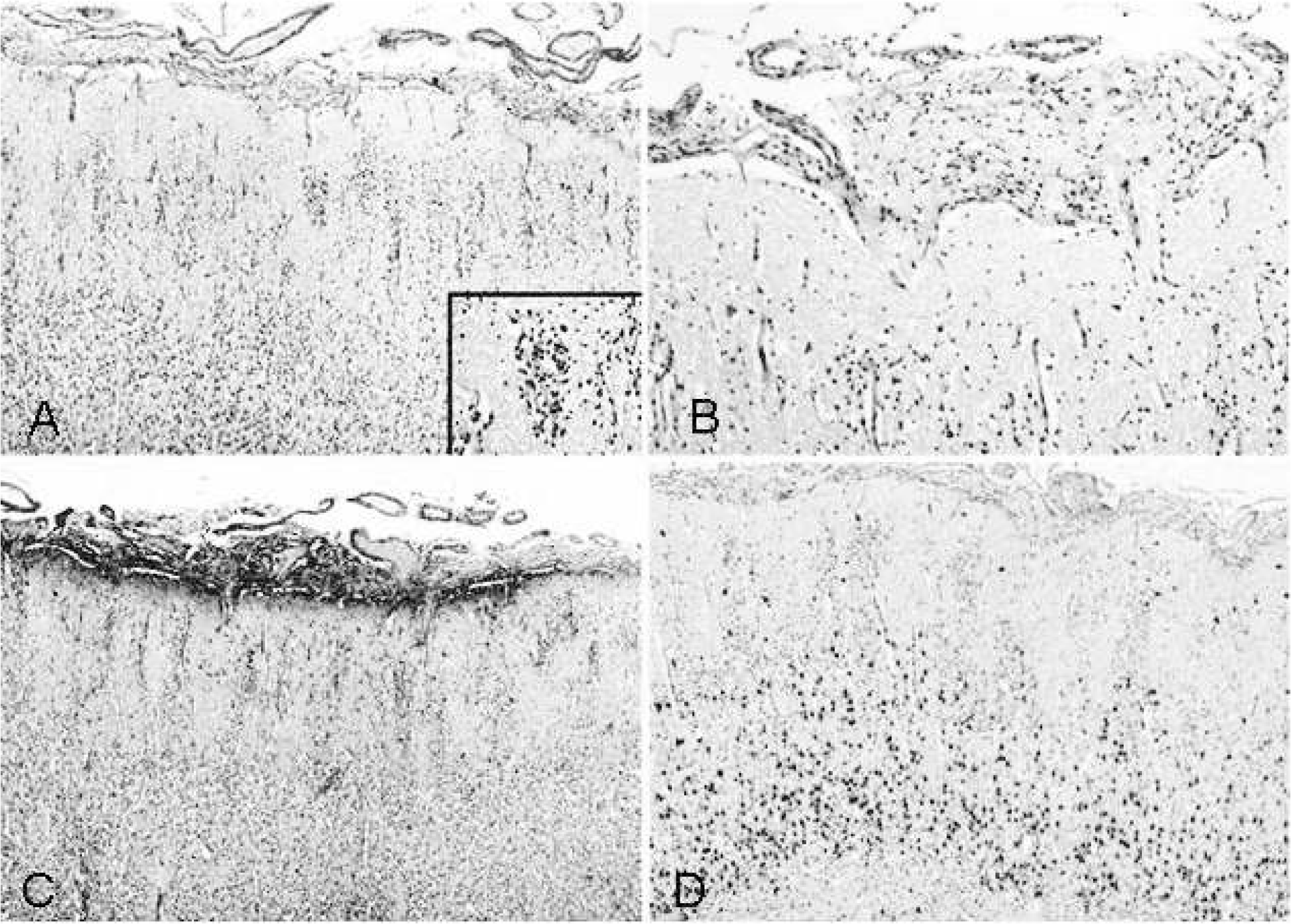
Histological features of the cortical alterations are shown. (
Cases of acardius with neuropathological features evaluated at autopsy (n = 40)

CONCLUSIONS
We report for the first time a case of acardius with features of acardius amorphus and acormus with a relatively well-differentiated brain with a minor form of holoprosencephaly (lobar holoprosencephaly), with neuropathologically disclosed intracerebral pigmented retina-like tissue, while optic pathways and eyeballs could not be detected. These findings call into question the current classification of acormus as well as the theory that insertion, that is, distances between the umbilical cord insertions, influences the acardiac morphology. Furthermore, it highlights the fact that we still know far too little about differentiation, pathophysiology, and genetics of acardiac twins, items essential to the establishment of new prognostic markers and therapeutic regimens. By investigating the mechanisms behind the TRAP sequence, we can learn more about the biology and pathology of this special form of monochorionic twin pregnancies.
Footnotes
ACKNOWLEDGMENTS
The authors thank Mr Mike Gerhardts for helping with the artwork and Mr Stefan Lampe for technical support. Written informed consent was obtained from the parents for publication of this case report and any accompanying images. A copy of the written consent is available on request.