
Abstract
Thymic involvement by Langerhans cell histiocytosis (LCH) has been described mainly in isolated case reports. A description of the histopathologic patterns of LCH proliferations in the thymus, together with therapeutic implications, has not, to our knowledge, been previously addressed. The pathology consultation files at Children's Hospital of Pittsburgh of UPMC were reviewed for cases of thymic involvement by LCH. Relevant cases in the literature were also reviewed, and the histopathology and clinical course of those cases were collected. Nine consultation cases of thymic involvement were reviewed, together with 23 cases in the literature, which provided adequate pathologic description and ancillary confirmation (n = 32), revealing 4 distinct pathologic groups. Group 1 showed microscopic collection of hyperplastic LCH-like cells in incidental thymectomies of patients without LCH disease, requiring no further treatment (n = 7; 22%). Group 2 showed solitary and/or cystic LCH of the thymus with gland disruption, and at least 3 cases resolved without systemic therapy (n = 10; 31%). Group 3 showed more variable thymic involvement in multi-systemic LCH disease, with either a medullary restricted pattern or more diffuse gland involvement, requiring adjuvant therapy and having a higher mortality rate (n = 13; 41%). Group 4 showed a mixed histiocytic lesion with a concurrent LCH and juvenile xanthogranuloma-like proliferation (n = 2; 6%). Thymic involvement in LCH is quite rare. Based on our cases and those in the literature, we propose 4 distinct pathologic groups of thymic involvement in Langerhans cell proliferations with relevance for diagnosis and treatment.
INTRODUCTION
Thymic involvement by Langerhans cell histiocytosis (LCH) has been described mainly in isolated case reports. Older studies [1–8] demonstrated diffuse thymic involvement in children with disseminated LCH who died of their disease. This older thymic LCH pathology literature, written before the advent of CD1a as a marker, is unreliable because macrophages could not be reliably distinguished from LCH cells and is omitted from further discussion here. Although thymic involvement by LCH is not new, until recently, there was little epidemiologic data on its incidence and effect on outcome. Two newer studies [9,10] support the notion that thymic involvement of multisystem (MS) LCH occurs at a younger age (0.7–2 years) and is associated with skin and high-risk involvement (ie, bone marrow, liver, spleen), along with higher rates of reactivation and mortality [10]. Both of these studies and isolated case reports highlight the importance of thymic involvement in MS LCH disease. However, there is little description in the literature [10–19] of the pathologic patterns of thymic involvement in the context of Langerhans cell proliferations and LCH disease.
Normal Langerhans cells (CD1a+/langerin+) are present in the thymic medulla as single cells or in association with the squamous epithelial Hassall corpuscles as dispersed, small, branched dendritic cells. Langerhans cell histiocytosis is best regarded as a myeloid cell neoplasm, which is likely derived from a bone marrow precursor cell [20]. Although both endogenous thymic Langerhans cells and LCH show Birbeck granules on ultrastructural examination and have an immunophenotype of CD1a and langerin positivity, LCH displays collections of larger, oval, nonbranched cells with convoluted nuclei [1,2,21,22]. In general, langerin (CD207) is the preferred marker for LCH in the thymus because small cortical thymocytes are CD1a+ (but langerin-), and staining for both markers provides diagnostic confidence (21). For the purposes of our discussion, we use the term hyperplastic LCH-like cells to refer to solitary aggregates of nonbranched, oval cells with convoluted nuclei (ie, LCH-like) that are histologically different from the endogenous, spindly dendritic cells of the thymus but do not cause gland destruction. We propose that there are actually 4 distinct clinicopathologic groups related to Langerhans cell involvement of the thymus with divergent therapeutic implications. The histopathology and clinical course of these groups are illustrated in the context of the current literature on LCH disease in the thymus.
METHODS
Review of Children's Hospital of Pittsburgh Cases
A review of pathology files from Children's Hospital of Pittsburgh, University of Pennsylvania Medical Center, 2000 to 2014, revealed 9 consult cases of thymic involvement by LCH-like hyperplasia and LCH from our clinical consultation files. There was no excess material available for molecular diagnostic or additional ancillary testing.
Review of Cases from the Literature
The literature searched for any combination of LCH and thymus included 122 cases. Cases published before 1987 and/or with limited pathologic-based data made it difficult to determine whether those cases met the 1987 Histiocyte Society histopathologic criteria of LCH [23]. Most of these cases are not included in this review, including those cases diagnosed by imaging studies (n = 62) (Supplemental Table 1; http://dx.doi.org/10.2350/15-01-1593-OA.S1). Only 23 of 122 cases (19%) in the literature mention the histologic features of thymic involvement with ancillary confirmation of the LCH cell (ie, ultrastructural or immunohistochemical study).
Group 1: Cases of incidental, microscopic Langerhans cell histiocytosis–like hyperplasia in the thymus
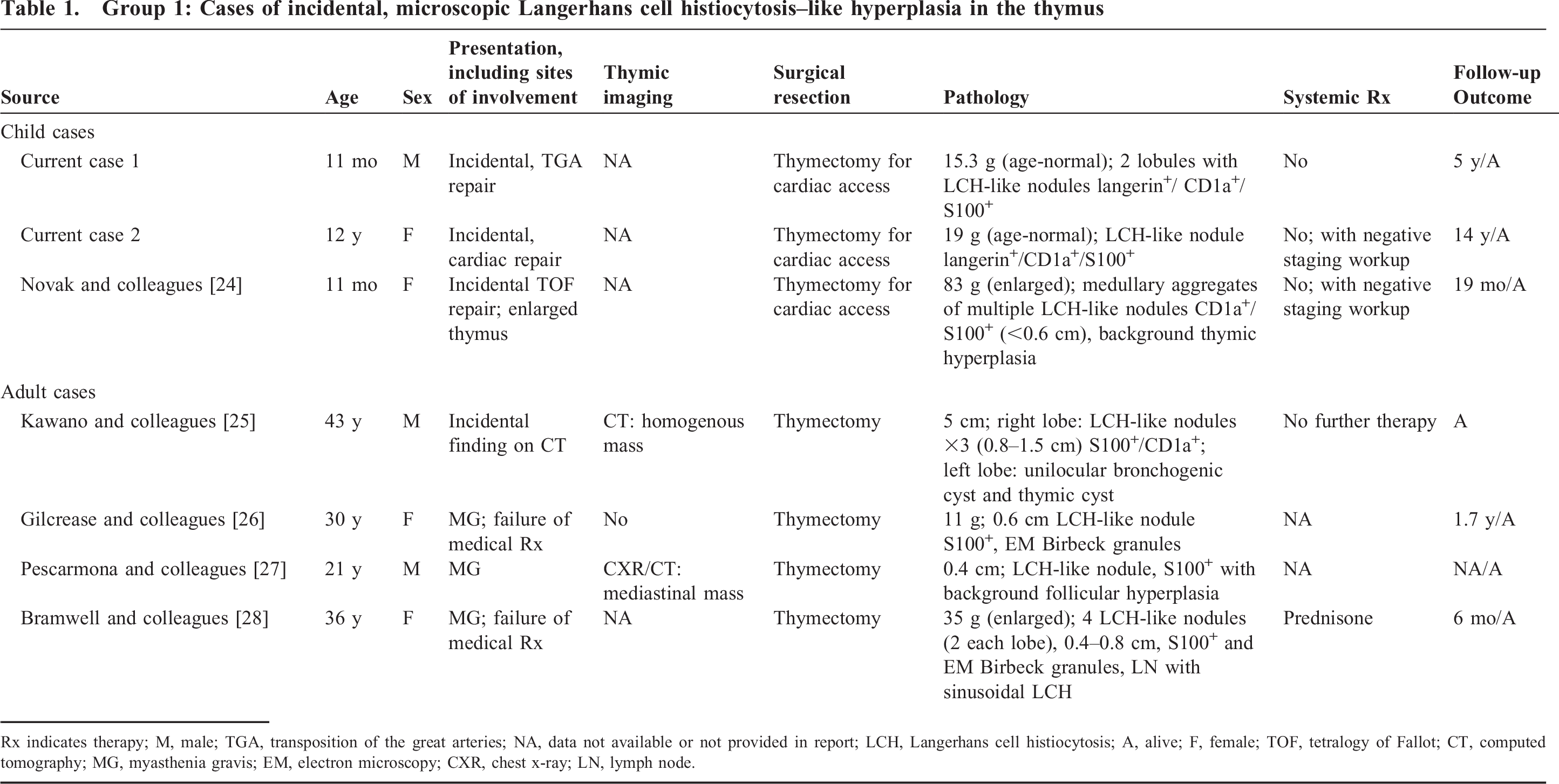
Rx indicates therapy; M, male; TGA, transposition of the great arteries; NA, data not available or not provided in report; LCH, Langerhans cell histiocytosis; A, alive; F, female; TOF, tetralogy of Fallot; CT, computed tomography; MG, myasthenia gravis; EM, electron microscopy; CXR, chest x-ray; LN, lymph node.
RESULTS
Thirty-two cases (n = 9 study cases and n = 23 cases from the literature with pathologic confirmation) delineate into 4 separable clinicopathologic groups of Langerhans cell proliferations within the thymus, which included group 1: incidental, LCH-like hyperplasia requiring no further treatment (Table 1); group 2: single-site (SS), solitary, or cystic LCH of the thymus with gland disruption and fibrosis that has the propensity to resolve without systemic therapy (Table 2); group 3: variable histologic thymic involvement, from confined medullary LCH aggregates to architectural disruption in the context of MS LCH, requiring systemic therapy and having worse survival rates (Table 3); and group 4: a mixed histiocytic lesion of LCH with concurrent juvenile xanthogranuloma (JXG)/reticulohistiocytoma (RH) type proliferation.
Group 2: Solitary Langerhans cell histiocytosis lesions of the thymus
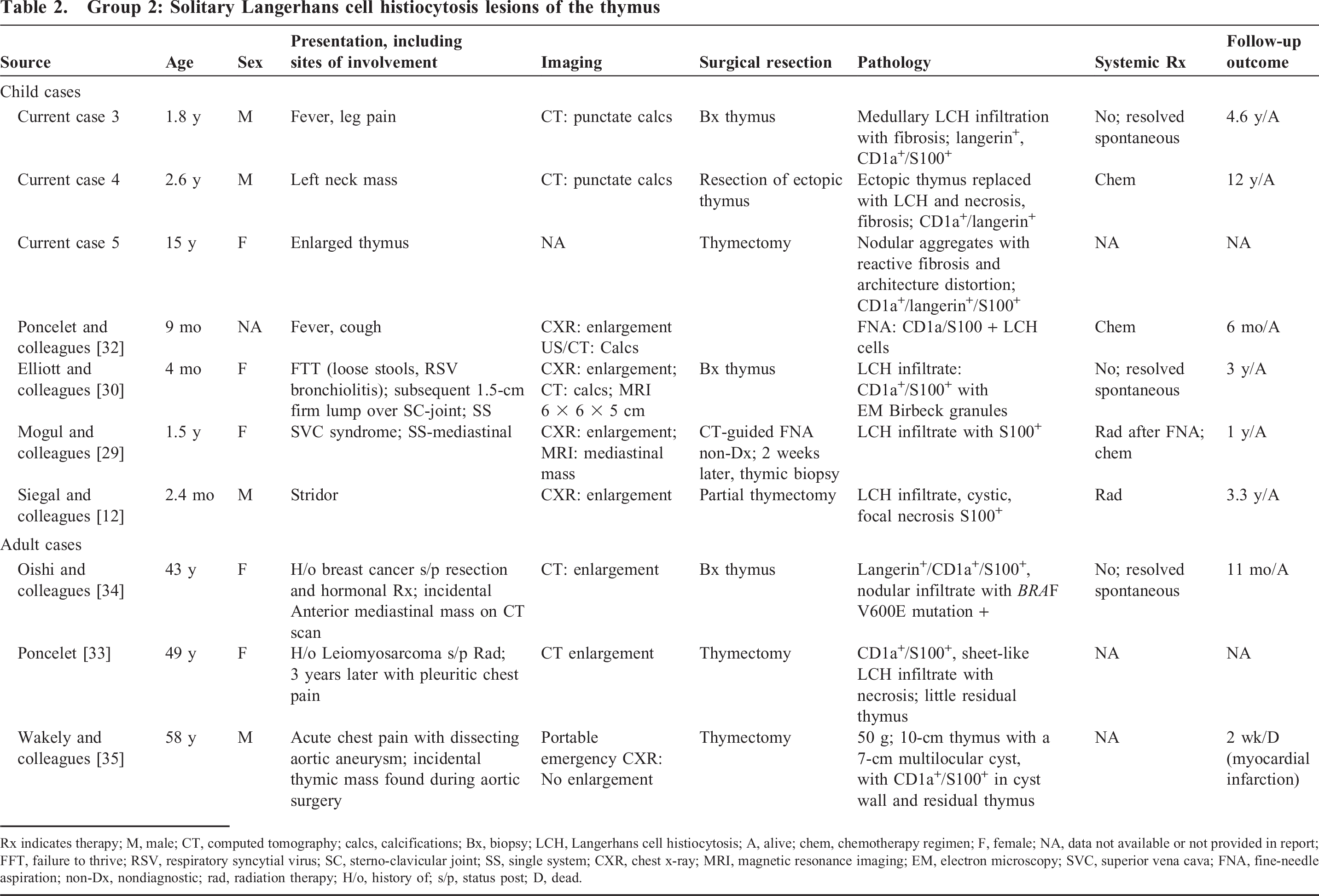
Rx indicates therapy; M, male; CT, computed tomography; calcs, calcifications; Bx, biopsy; LCH, Langerhans cell histiocytosis; A, alive; chem, chemotherapy regimen; F, female; NA, data not available or not provided in report; FFT, failure to thrive; RSV, respiratory syncytial virus; SC, sterno-clavicular joint; SS, single system; CXR, chest x-ray; MRI, magnetic resonance imaging; EM, electron microscopy; SVC, superior vena cava; FNA, fine-needle aspiration; non-Dx, nondiagnostic; rad, radiation therapy; H/o, history of; s/p, status post; D, dead.
Group 3: Thymic involvement in multisystem Langerhans cell histiocytosis disease
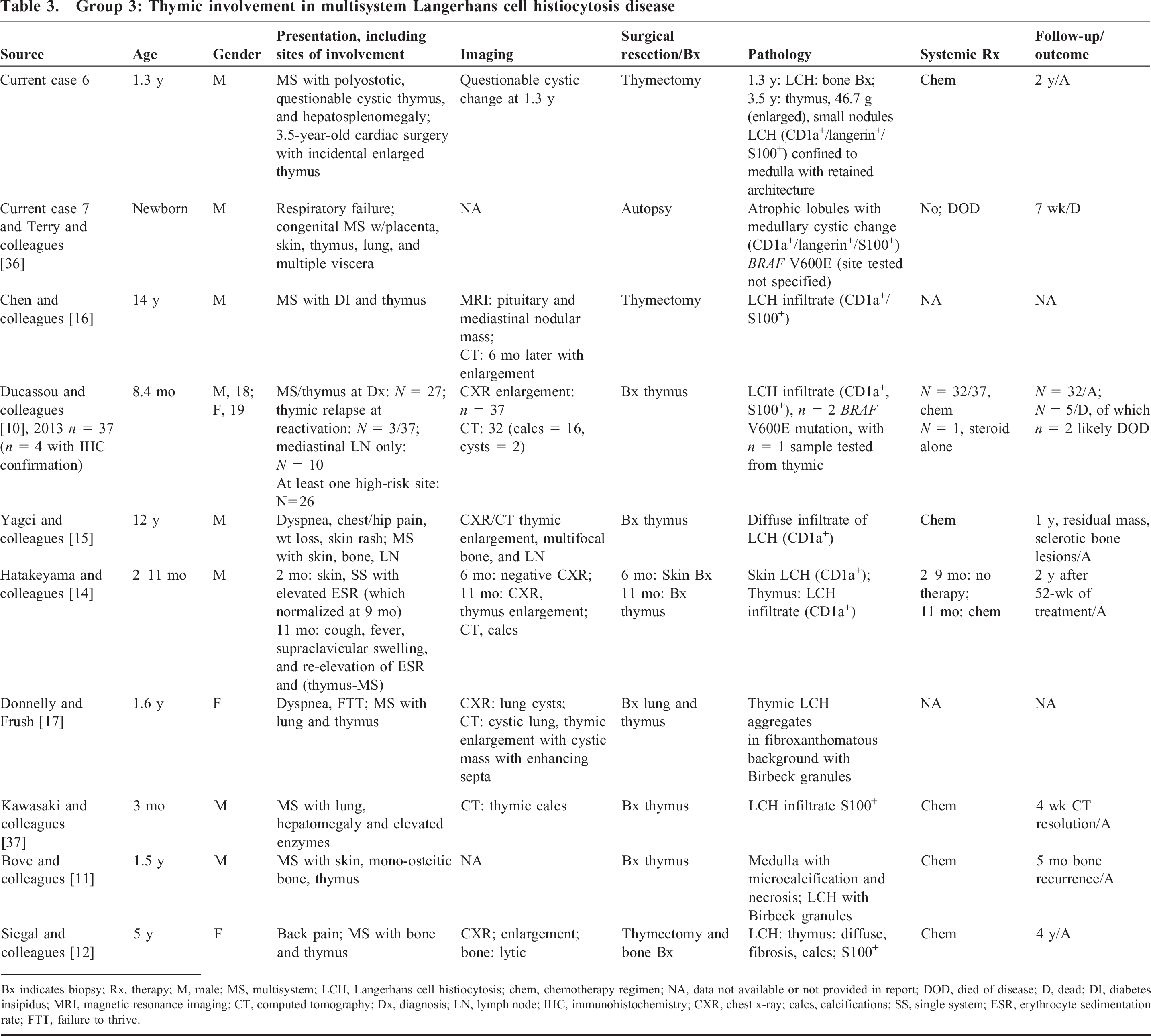
Bx indicates biopsy; Rx, therapy; M, male; MS, multisystem; LCH, Langerhans cell histiocytosis; chem, chemotherapy regimen; NA, data not available or not provided in report; DOD, died of disease; D, dead; DI, diabetes insipidus; MRI, magnetic resonance imaging; CT, computed tomography; Dx, diagnosis; LN, lymph node; IHC, immunohistochemistry; CXR, chest x-ray; calcs, calcifications; SS, single system; ESR, erythrocyte sedimentation rate; FTT, failure to thrive.
Group 1: Incidental LCH-like hyperplasia
Study cases
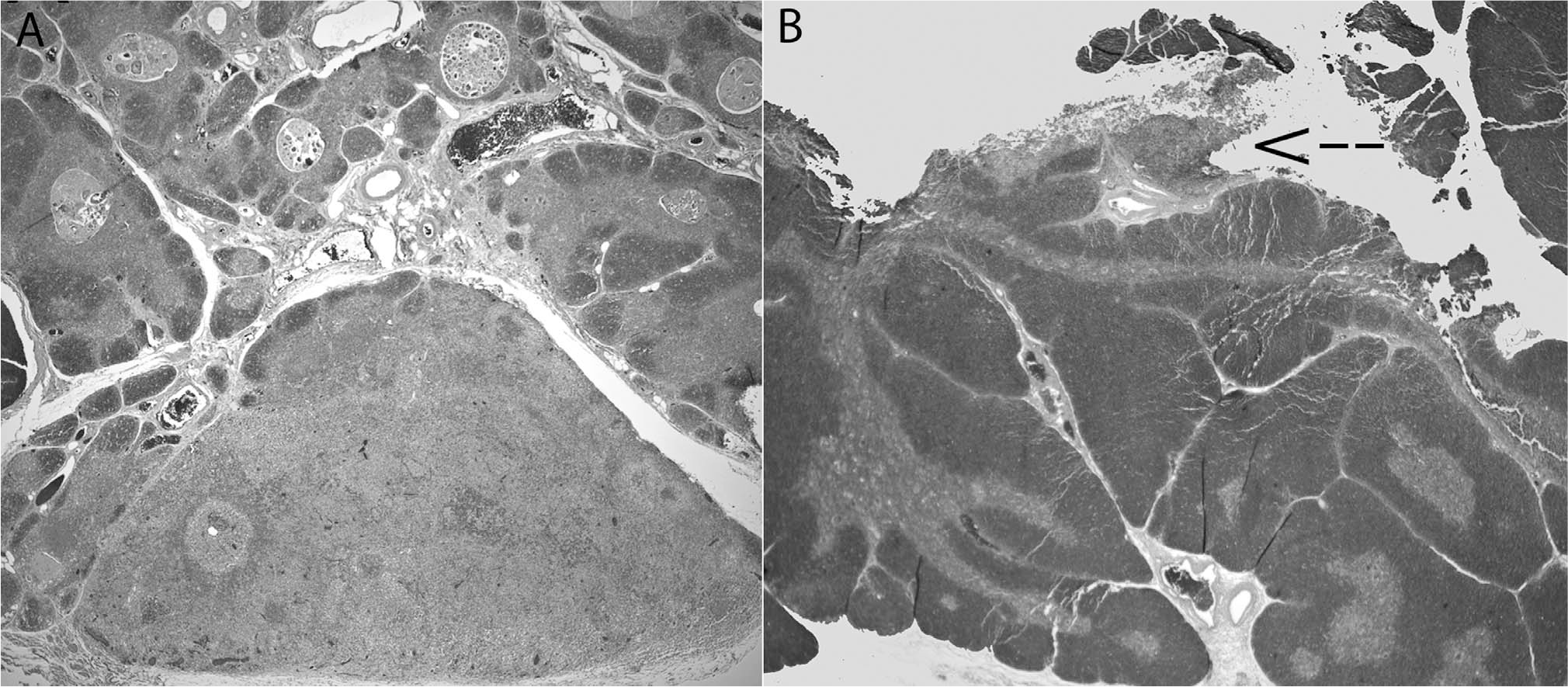
Group 1: Incidental Langerhans cell histiocytosis (LCH)–like hyperplasia.
Review of cases, including those from the literature
Five additional cases were found in the literature with small, incidental, LCH-like, microscopic hyperplasia of the thymus and are outlined in detail in Table 1. In all, 7 patients with LCH-like hyperplasia were found during incidental thymectomy or as an incidental finding during unrelated computed tomography (CT) imaging [24–28]. None had evidence of LCH disease, either at the time of diagnosis or in subsequent follow-up. The histopathology showed a small nodule or single lobule replaced by a collection of hyperplastic LCH-like cells with oval cytoplasmic contours and a characteristic “coffee-bean” nucleus with interspersed eosinophils. The background thymic corticomedullary architecture was largely intact without evidence of fibrosis, but some showed dilated Hassall corpuscles. There was a bimodal age distribution, with younger age (mean [SD], 4.6 [6.4] years) in those patients undergoing thymectomy for cardiac access as compared with adults with thymectomy (mean [SD], 32.5 [9.3] years). There was no particular gender preference (3 males, 4 females). Only 3 adult patients had a history of myasthenia gravis (MG), and there was no reported history of lymphoma in any of these cases (Table 1).
Group 2: Solitary (unifocal) tumoral and/or cystic thymic SS LCH
Study cases
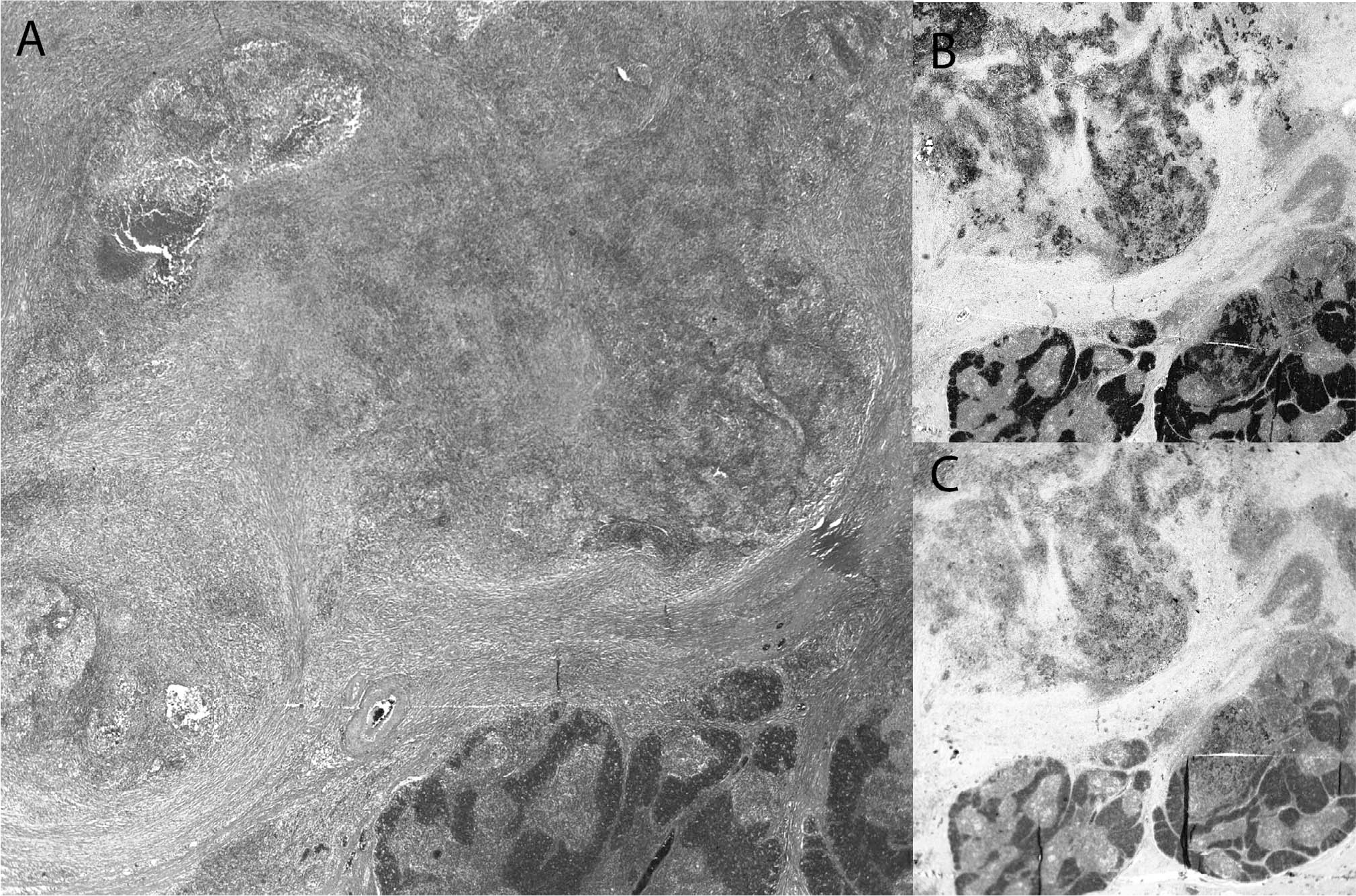
Group 2: Solitary mediastinal mass in single-site Langerhans cell histiocytosis (SS-LCH).
Review of cases, including those from the literature
The SS LCH of the thymus differs from the incidental LCH-like hyperplasia largely in the extent of the process within the thymus, based on gland destruction with necrosis and punctate calcifications and a surrounding fibrotic response. The 11 cases from our literature review (including our 3 cases) that fall into this group are outlined in detail in Table 2 [12,29–35]. There was a bimodal age distribution of young patients (mean [SD], 3.5 [5.3] years) and older patients (mean [SD], 50 [7.5] years), without a significant gender predilection (8 females, 5 males). Anterior mediastinal widening or mass, either on chest x-ray, CT scan, and/or magnetic resonance imaging (MRI) was the most frequent description, whereas punctate calcifications by CT and/or ultrasound were also common. None of these cases had a history of MG or lymphoma.
Group 3: Multisystem LCH with thymic involvement
Case study
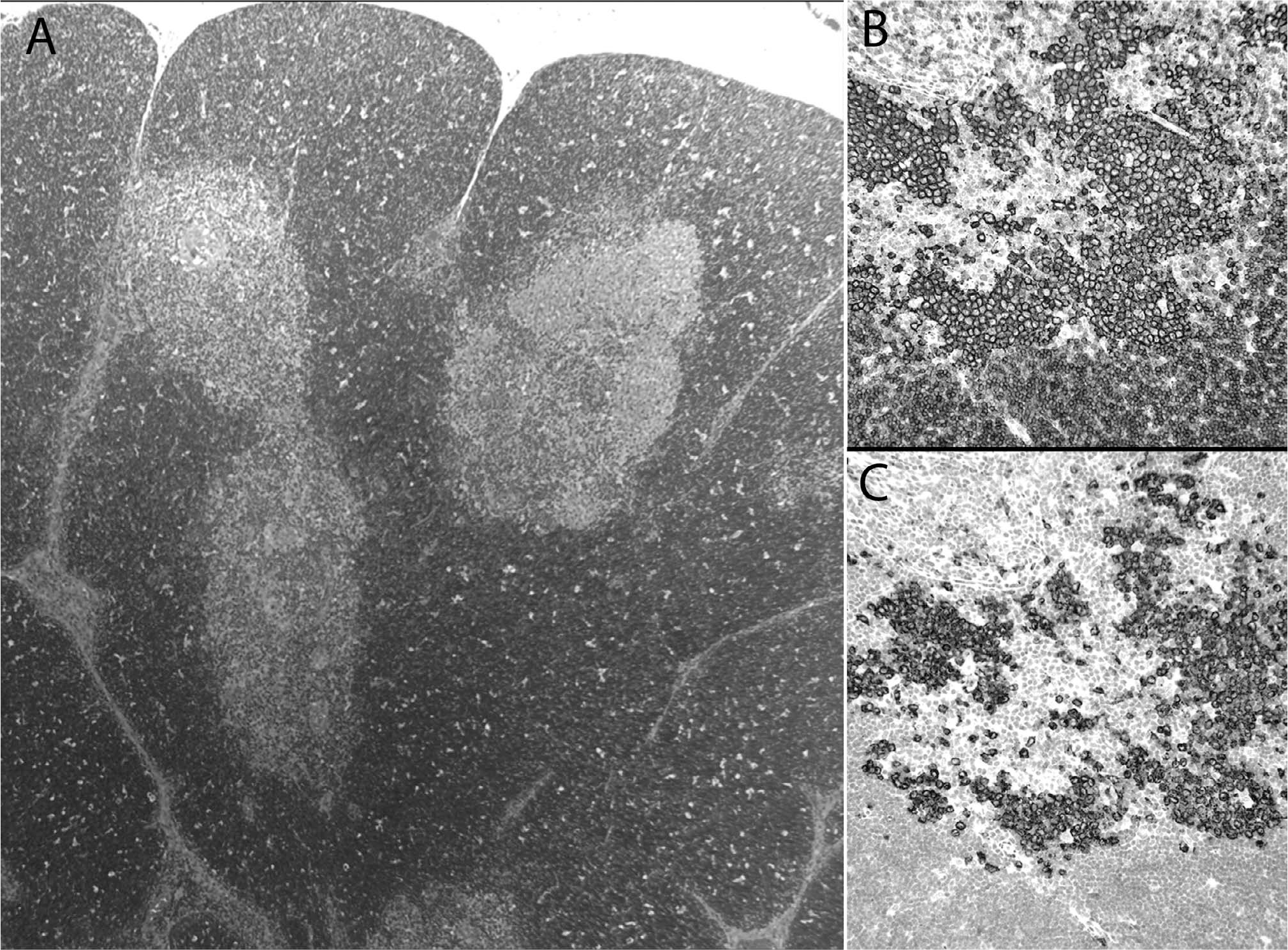
Group 3: Multisystem Langerhans cell histiocytosis (MS-LCH).
Review of cases, including those from the literature
Involvement in MS LCH represents the largest group of thymic LCH characterized in the literature. However, although our review identified numerous reported cases of thymic involvement in the context of MS LCH (Supplemental Table 1; http://dx.doi.org/10.2350/15-01-1593-OA.S1), only 11 cases [10–12,14–17,37] and our 2 consult cases (Table 3) had ancillary confirmation of LCH in the thymus. Of note, a case of a fibrosing mediastinitis in the context of bone LCH may represent a “burnt-out” type LCH pattern with the absence of LCH cells in the thymus (CD1a- and S100-) with a fibrotic background; however, for the purposes of this study, that case was not included in the cohort [18]. This group showed a predilection for younger children (mean [SD], 3.2 [5.3] years) with a skewed gender distribution of males (n = 9) compared with females (n = 2); however, gender could not be ascertained in 4 cases [10]. Although many MS LCH cases in the literature do not describe the histologic pattern of thymic LCH involvement, our cases, together a few reports with ancillary confirmation describe a variable histologic appearance that included confined medullary involvement to diffuse gland involvement with fibrosis (Table 3). Of note, 3 cases harbored the BRAF V600E mutation, in which one demonstrated a thymic LCH infiltrate [10,36]. In addition, none of these cases had a history of MG or lymphoma.
Group 4: Mixed histiocytic lesions with LCH and JXG/RH proliferation
Case study
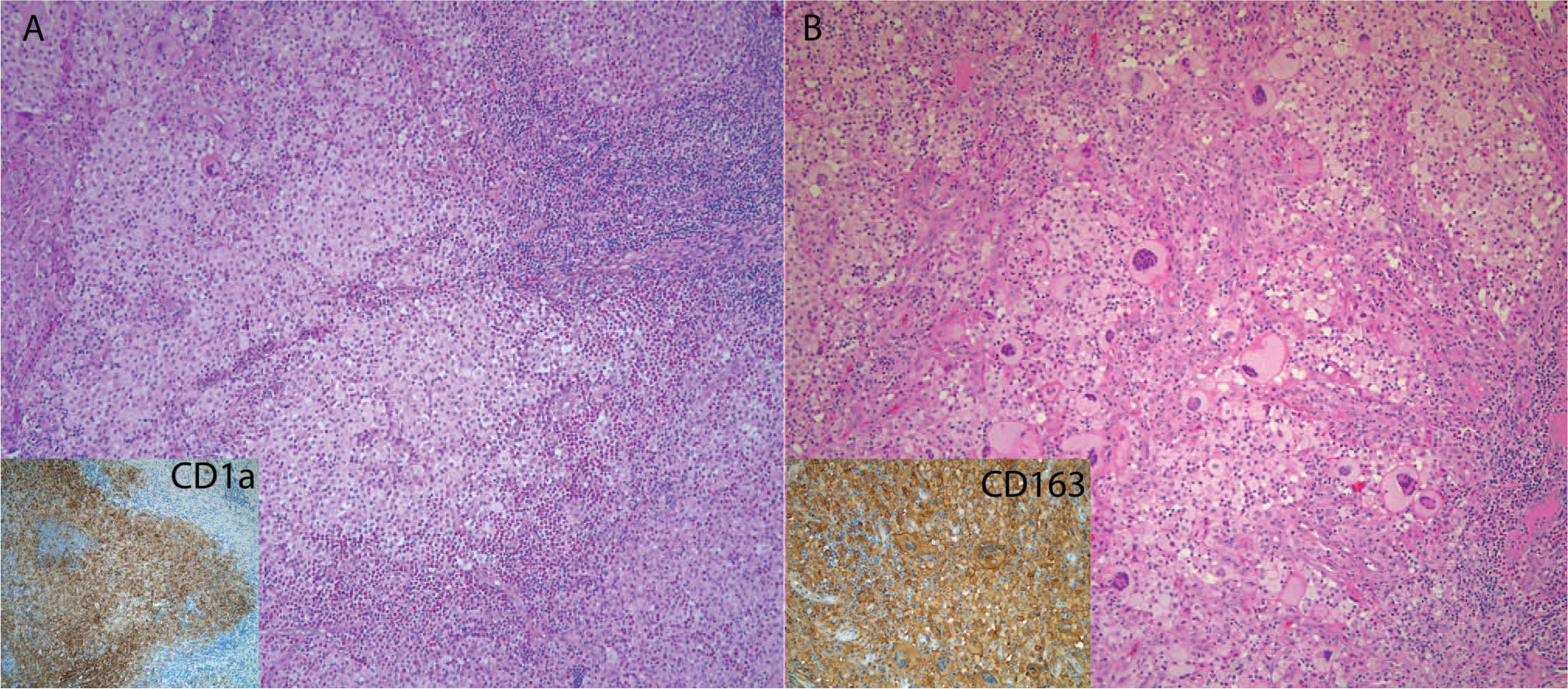
Combined juvenile xanthogranuloma and Langerhans cell histiocytosis (LCH).
Following her surgery and a diagnosis of LCH, a complete skeletal survey did not show evidence of bone lesions, and liver function tests were within reference range. The CT scan of the chest a few months after diagnosis showed a residual mediastinal mass and otherwise clear lung parenchyma. A repeat CT scan 9 month after diagnosis showed a stable-appearing mediastinal mass, indistinguishable from thymus. At the 1-year follow-up, she was alive and well without systemic therapy. Neither of these patients had a history of MG or lymphoma.
DISCUSSION
We describe an illustrative clinicopathologic case series with review of the pertinent literature to demonstrate 4 pathologic patterns of thymic involvement in LCH disease or LCH-like proliferations with distinct therapeutic implications.
We propose that the pathology of LCH-like hyperplasia is distinct from LCH disease, suggesting that this pattern of thymic involvement represents a distinctive population, likely reactive in nature. This pattern has been previously noted by others in incidental thymectomies [24–28]. Christie and colleagues [38] described LCH-like proliferations and demonstrated that some were polyclonal, unlike LCH. We think it is important to separate these incidental, small lesions from LCH because they do not appear to occur outside of an SS, nor do they progress clinically, but they do cause confusion when diagnosed as LCH. The mechanism of proliferation and etiology of the cells is not currently known, but they appear different from the endogenous Langerhans cell population of the thymus that display spindled dendritic cell processes. Although the possibility of an early LCH cannot be entirely excluded, these nodules are found incidentally and are small, without the type of gland destruction that is seen in SS LCH of the thymus (vide infra). Foci of reactive, LCH-like proliferations with plump, round cells have previously been described in lymph nodes harboring other histiocytic lesions, such as Rosai-Dorfman disease, lymphomas/leukemia, and in association with soft tissue–treated tumors (ie, rhabdomyosarcoma and melanoma) [38]. An altered immunologic milieu was suggested as a driver for reactive and proliferate, LCH-like hyperplasia in these cases [38]. Of note, in 1984, Halícek and Rosai [39] described in myasthenic patients a series of thymuses with a reactive hypereosinophilic appearance similar to the reactive eosinophilic pleuritis seen in patients with spontaneous pneumothoraces. The pathology was described as nodular aggregates of histiocytes and eosinophils (ie, histioeosinophilic granulomas); however, unlike our cases, they could not demonstrate morphologic, immunohistochemical, or ultrastructural confirmation of Langerhans cells [39]. Our current case series is limited because we were unable to further assess clonality or BRAF mutation status in these cases. Patients in this group did not show evidence of LCH disease. Although one adult patient with MG from the older literature was treated with steroid therapy [28], most did well without the need for systemic therapy.
The histopathology of solitary thymic LCH has been not been previously described with detail in the literature. In SS LCH of the thymus, the architecture is largely replaced with an LCH infiltrate presenting as a solid or cystic mediastinal mass. That infiltrate forms nodular tumors comprising LCH cells with areas of punctate calcifications and/or foci of necrosis and a loose edematous to dense fibrotic response, which disrupts the architecture of the gland. Workup for LCH disease limited to the thymus includes diagnostic staging to exclude concomitant sites of involvement and to establish the disease as an SS LCH [19]. The use of systemic therapy seems to be variable and may be influenced by concern about airway compromise (31). Treatment with either radiation therapy [12,29] and/or chemotherapy [29,32] is more often described in the older literature of thymic SS LCH. Although this small case series and review of the literature cannot make definitive claims for treatment, there is at least the suggestion that thymic SS LCH has the propensity to undergo spontaneous regression (after biopsy or aspiration cytology) without systemic treatment, as demonstrated in case 3 and others in the literature [30,34]. Solitary, thymic LCH involvement may be similar to other instances of SS LCH in the bone, skin, and lymph nodes, in which an initial approach of close follow-up without systemic therapy may be the treatment of choice in the absence of other lesions after diagnostic staging. However, prospective studies are needed to confirm this conclusion.
Thymic involvement of MS LCH occurs predominately in the youngest (<1 year) patients with LCH, both with and without systemic symptoms, but all with active disease [9,10], which was also seen in our study cases. Thymic involvement may not be readily recognized at the time of LCH diagnosis if not directly imaged. Newer studies suggest that routine screening of young patients with LCH may be prudent in the initial staging evaluation, especially given the association with more high-risk involvement and propensity for reactivation in these youngest patients [9,10]. The presence of thymic enlargement, intrathymic punctate calcification, and/or cystic transformation all have been described as diagnostic radiographic features of involvement (Supplemental Table 1; http://dx.doi.org/10.2350/15-01-1593-OA.S1) [9,12–14,31,40–45]. In the literature, there have been about 32 reported cases of thymic involvement by imaging with associated LCH high-risk involvement of either the spleen, liver, or hematopoietic system; however, most cases did not have histologic confirmation of thymic involvement [10,46,47]. Ducassou and colleagues [10] have shown that mediastinal LCH, in the context of MS LCH, tends to have higher rates of reactivation and mortality, as compared with nonmediastinal MS LCH [10]. Unlike solitary thymic LCH, thymic involvement in the context of MS LCH may have a more-aggressive natural history. However, it remains unclear whether thymic involvement in MS LCH is an independent risk factor or merely related to these cases tending to occur in younger children with more associated high-risk organ involvement.
Lastly, there are reports in the literature of mixed histiocytic disorders either occurring sequentially or concurrently, in the same sites or in different locations. These include JXG lesions occurring after LCH [48,49] and concurrent LCH and Erdheim-Chester Disease (ECD) or ECD following LCH [50]. However, there have been no previous reports, to our knowledge, of concurrent LCH and JXG-family of lesions occurring in the thymus. Our 2 cases are both from young patients. One case had MS LCH with high-risk involvement (case 8), whereas the other case had apparent solitary thymic LCH that required no further treatment (case 9); however, the presence of a mediastinal mass after thymectomy in case 9 remains a concern. The therapeutic implications of mixed histiocytic disorders in the thymus are not well delineated, given the few cases, but should be at least be followed with complete clinical staging to rule out MS LCH involvement.
In conclusion, we describe 4 distinct histologic patterns of Langerhans cell proliferations and LCH of the thymus with relevance for diagnosis and treatment.
Future work exploring the clonality and molecular status of these lesions is needed to better understand the pathophysiology and biologic potential of these cells, especially in cases of incidental LCH-like proliferations and mixed histiocytic disorders.
Footnotes
Conflict of interest statement: The authors report no conflict of interest.