
Abstract
Congenital adrenal agenesis is an extremely rare condition wherein the adrenal glands fail to develop. The absence of adrenal tissue results in the complete absence of hormones produced in the adrenal cortex (cortisol, aldosterone) and medulla (catecholamines), and is not compatible with postnatal life without artificial hormone replacement therapy. To date, 9 cases of adrenal agenesis have been reported, many of which are associated with additional congenital anomalies. Most cases were not detected on antenatal imaging and were detected incidentally at postmortem examination. We present a case of adrenal agenesis, detected incidentally at postmortem examination after termination of pregnancy for suspected fetal hydrops, and review the heterogeneous phenotype of this condition with associated abnormalities and molecular genetics. This case reinforces the role of the perinatal autopsy to investigate cause of perinatal mortality, allowing correlation of pathology with antenatal imaging findings and clinical details.
INTRODUCTION
Congenital adrenal agenesis is an extremely rare condition wherein the adrenal glands fail to develop. This condition differs significantly from adrenal hypoplasia or congenital adrenal hyperplasia, conditions in which the adrenal glands are present, but do not function adequately. In adrenal agenesis, the absence of adrenal tissue results in the complete absence of hormones produced in the adrenal cortex (cortisol and aldosterone) and medulla (catecholamines), and, thus, is not compatible with postnatal life without artificial hormone replacement therapy.
CASE REPORT
We present the case of a stillborn female infant of nonconsanguineous parents from rural Australia, delivered at 24 weeks of gestation to a previously healthy 20-year-old primigravida. Ultrasound at 12 weeks of gestation suggested normal fetal morphology. Combined first trimester screening revealed low risk for Trisomy 18 and 21. The pregnancy was uncomplicated until the routine morphology ultrasound scan at 19 weeks of gestation demonstrated pericardial effusion and an echogenic left kidney. Referral was made to a specialist perinatal ultrasound clinic and repeat study suggested no fetal abnormality. Further ultrasonography at 21 weeks of gestation demonstrated oligohydramnios, an enlarged heart, and poor lung development with marked pericardial and pleural effusions, and bilateral renal agenesis. Following genetic counseling, a decision was made to terminate the pregnancy. After induction of labor, a stillborn female infant was delivered, weighing 440 g.
The infant's parents consented to full postmortem examination.
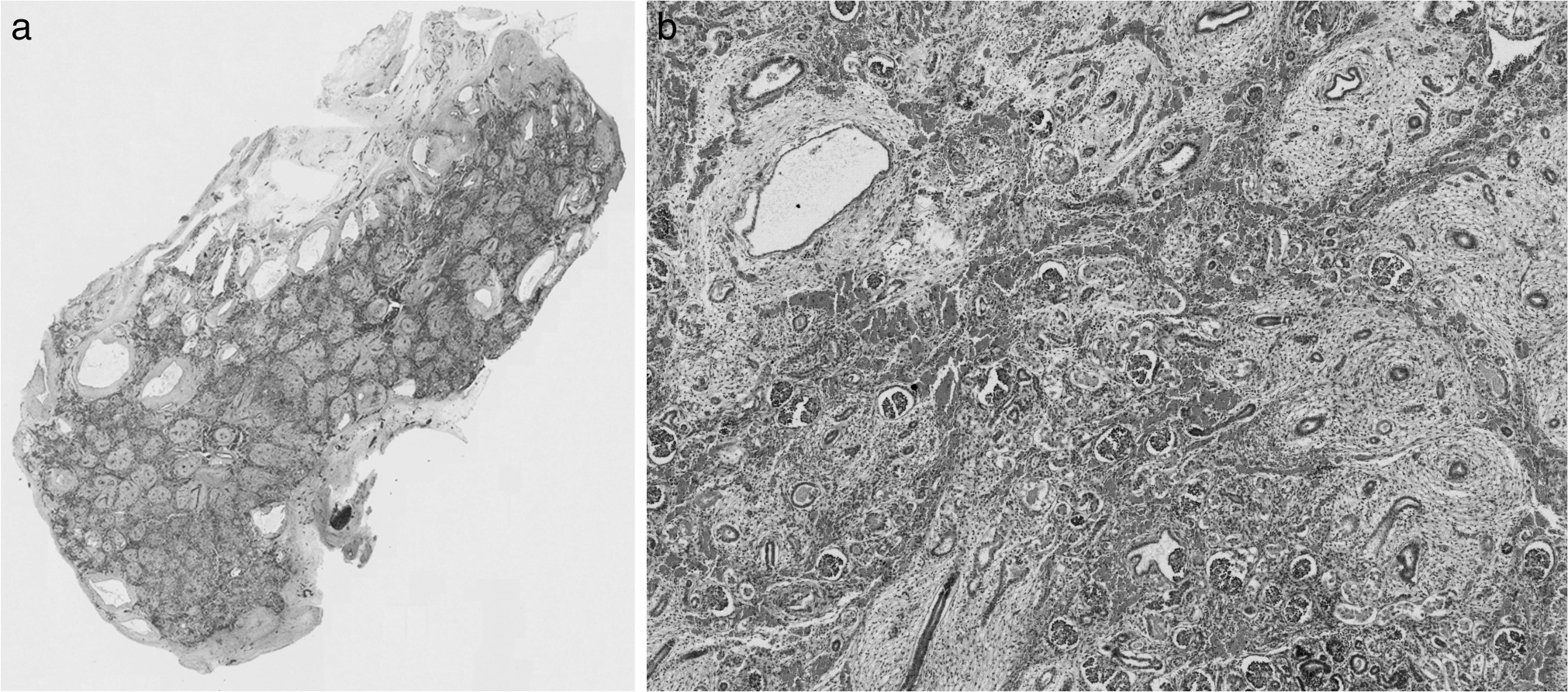
External examination revealed a well preserved, dysmorphic, female infant with organ weights and measurements below the 5th percentile for 24 weeks of gestation, and more in keeping with weights and measurements expected at 21 weeks of gestation. The growth restriction was harmonious with a brain-to-liver weight ratio of 4.0. The infant was noted to have a broad nose, large, flat rudimentary ears, micrognathia, retrognathia, and broad set eyes (Fig. 1A,B). Mild nuchal edema (Fig. 1B), and a narrow and bell-shaped chest also were present. There were no limb contractures or pterygia. Normal female genitalia were present. A significant finding on internal examination was the absence of adrenal glands bilaterally. This was confirmed macroscopically and histologically. Additional postmortem findings confirmed on postmortem skeletal survey and MRI included thymic hypoplasia (thymus weight 0.35 g, normal 5th–95th percentile 0.61–3.03) [1] and mild pulmonary hypoplasia bilaterally (right lung weight 4.08 g, normal 5th–95th percentile 5.52–13.06 g; left lung weight 2.7 g, normal 5th–95th percentile 4.74–10.71 g) [1]. A cystically dysplastic left kidney also was noted (Fig. 2A,B); however, the right kidney was present and normally formed. There was no hydronephrosis. The left ureter was 1.5 mm in diameter, narrow but probe patent. The right ureter was of normal caliber and patent. The bladder contained approximately 5 mL of clear urine and the urinary system otherwise was unremarkable. Bilateral ovaries and the uterus were identified in the pelvis, the pituitary was located in the pituitary fossa, and the central nervous, cardiovascular, endocrine, and gastrointestinal systems were unremarkable. No significant findings were detected on examination of the placenta.
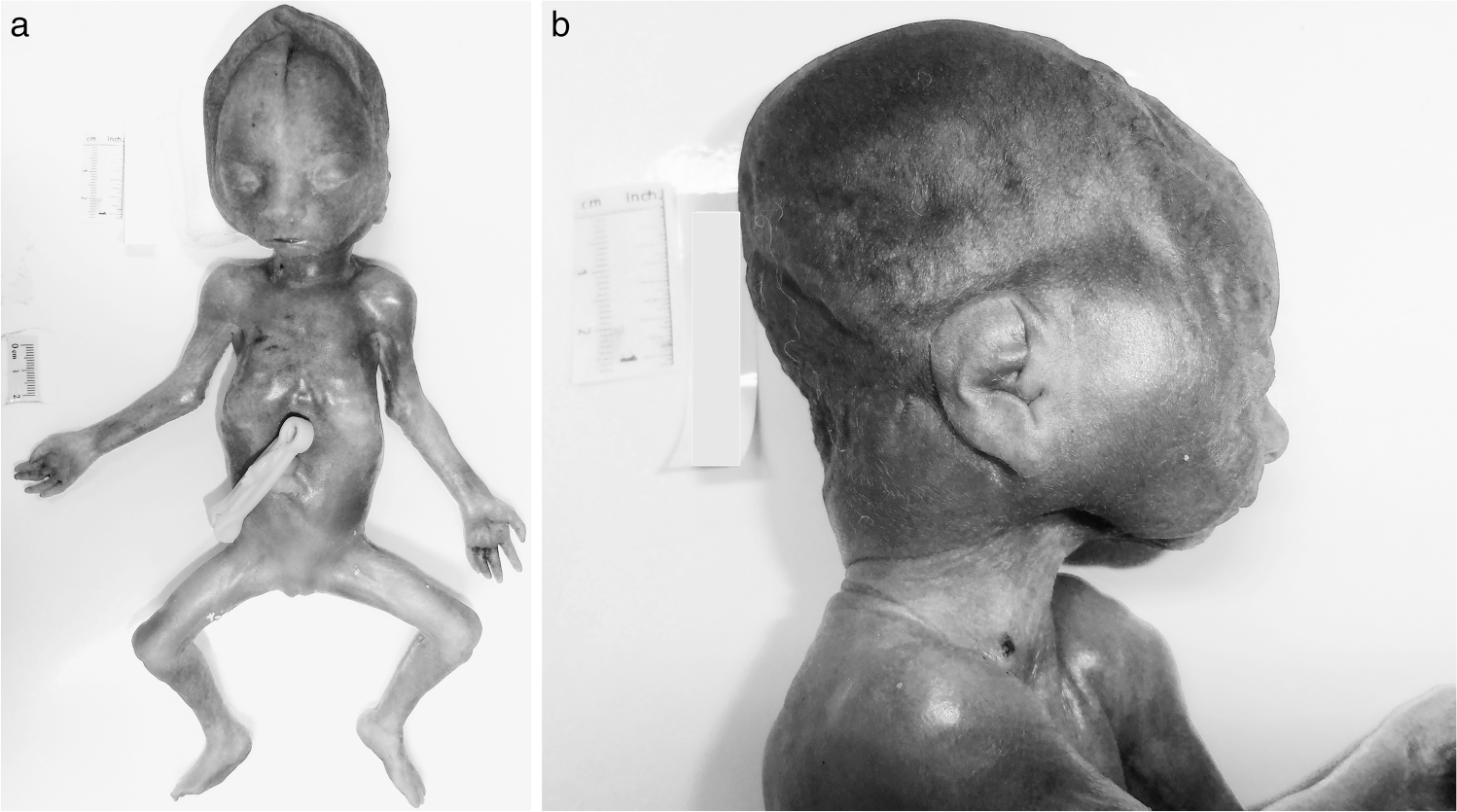
Dysmorphic female infant with absent adrenal glands.
Chromosome microarray analysis (CMA) was performed on DNA extracted from fetal rib cartilage using an Illumina HumanCoreExome-12 v1.0 microarray (Illumina, San Diego, CA, USA) and revealed a female molecular karyotype with an interstitial microduplication of approximately 1.3 Mb from chromosome region 16p13.11 (hg19 chr16:14,975,292–16,297,272×3). This well-documented recurrent microduplication [2,3] also was detected in the infant's father. The parents were referred for additional genetic counseling.
A single etiology for the external dysmorphic features, bilateral adrenal agenesis, left renal cystic dysplasia, and thymic hypoplasia detected at postmortem in this case could not be determined. The association with dysmorphic features in the absence of limb contractures and pterygia associated with severe oligohydramnios, suggests a syndromic phenotype. However, a specific genetic cause could not be identified using CMA analysis at an effective resolution of approximately 0.1 Mb.
DISCUSSION
Although congenital adrenal cortical hypoplasia is well documented, the true incidence and pathogenesis of congenital adrenal agenesis is unknown and information relating to pathogenesis is limited. To our knowledge, only nine cases of congenital bilateral adrenal agenesis have been reported previously to date [4–9], leaving some to suggest that the condition does not exist and, instead, is a result of poor dissection technique at postmortem. Additional reports of congenital unilateral adrenal agenesis have been reported, often associated with adrenal hypoplasia of the remaining adrenal gland. Adrenal aplasia and hypoplasia have been detected in association with anencephaly and pituitary disorders in humans [5]. Although adrenal agenesis is rare, there have been two reports of adrenal agenesis occurring in male siblings, suggesting a possible X-linked genetic mechanism in the pathogenesis of congenital adrenal gland disorders [6,9].
The clinical presentation of adrenal agenesis is heterogeneous and includes fetal death in utero and in liveborns, respiratory distress, pulmonary hypertension, and circulatory insufficiency immediately after birth relating to the absence of cortisol, catecholamines, and aldosterone [4–9]. Many of the reported cases of adrenal agenesis have additional anomalies, including renal, lung, spleen, and blood vessel anomalies. One reported case was associated with maternal diabetes [8]. The majority of reported cases of adrenal agenesis were not diagnosed on antenatal imaging, were not associated with oligohydramnios, and were detected incidentally at postmortem examination [6,8,9]. The reasons for the difficulty achieving a prenatal diagnosis of adrenal agenesis may include anatomic variation, the retroperitoneal suprarenal location of the adrenal glands, a lack of knowledge of the condition, and/or complications of other associated anomalies, including poor visualization in the context of oligohydramnios secondary to associated renal anomalies. Improving the antenatal detection rate of adrenal agenesis would enable lifesaving treatment to be given after birth and may prevent neonatal death.
The adrenal gland consists of the adrenal cortex, derived from the intermediate mesoderm, and the adrenal medulla, originating from the truncal neural crest cells that migrate under the influence of trophic factors, including adrenocortical steroids [10]. Thus, the development of the adrenal cortex and medulla are intimately related, and the adrenal medulla fails to develop in the absence of adrenal cortical tissue. This explains why both adrenal components were absent in our case. Improved understanding of embryology and adrenal developmental biology has led to the detection of potentially pathogenic gene mutations associated with adrenal agenesis. Mutations in the Wt1 gene, encoding the transcription factor Wt1, which is expressed in the coelomic epithelium of the urogenital ridge, results in adrenal agenesis together with gonadal agenesis in mice [11,12]. This is not surprising given both organs derive from the coelomic epithelium of the urogenital ridge. Additionally, in one case a 14-month-girl had bilateral adrenal agenesis with normal uterus and ovaries, and proved to have a heterozygous mutation in an orphan nuclear receptor steroidogenic-factor 1 (SF-1). SF-1 has a vital role in the development of the adrenal glands, as well as the gonads, pituitary gonadotropes, and hypothalamus [4]. Mice with SF-1 mutations also exhibit absent adrenals and gonads associated with hypothalamic and pituitary disease [13]. However, in one of the most recently reported cases of congenital adrenal agenesis, detected by intra-abdominal ultrasound soon after birth in a term liveborn female infant, none of the previously reported SF-1 mutations were detected [7]. Interestingly, the adrenal cortex and kidney share a number of molecular developmental pathways, including Wt1 and WNT4, and the presence of unilateral renal cystic dysplasia and one normal kidney in our case could represent an abnormal mesenchymal anlage or a form of haploinsufficiency [11,12]. Given that adrenal development is complex and involves at least 69 genes encoding multiple transcription factors, signaling molecules, steroidogenic hormones, and extracellular matrix proteins, it is likely that more than one gene is involved in the pathogenesis of adrenal agenesis [14].
A number of genes of interest regarding adrenal agenesis have been identified. In our case, molecular analysis was restricted to CMA analysis. The 16p13.11 microduplication detected in this infant contains in excess of 20 genes, including the nuclear distribution protein nudE homolog 1 (NDE1) gene, a gene expressed in 115 organs, including the adrenal cortex. While some reports suggest the 16p13.11 duplication is a risk factor for neurodevelopmental disorders and congenital abnormalities, others suggest it could be a benign copy number variant [2,3]. A recent study has found no significant difference in the frequency of the duplication between patients referred with intellectual disability and congenital defects when compared to unaffected controls [15]. Therefore, this duplication likely represents a benign variant or a susceptibility locus and bears no pathogenic relationship with the phenotype presented in this case. The duplication may be inherited from a healthy parent, and in this case was inherited from the father who had an unremarkable medical history.
The presence of unilateral renal cystic dysplasia in an otherwise normal urogenital system was surprising in our case, given the antenatal ultrasound findings suggested bilateral renal agenesis. The unilateral left ureteric narrowing implies obstructional unilateral cystic dysplasia. However, the presence of a normal right kidney excluded urogenital disease as a cause of oligohydramnios. Although a specific cause for the oligohydramnios was not identified in our case, it is possible that an undetected factor may have contributed, such as maternal dehydration or a fetal swallowing or neurological problem. The absence of adrenal glands is unlikely to have significantly contributed to the oligohydramnios: fetal aldosterone-controlled renal tubular reabsorption of sodium and water is maternally derived until the definitive fetal adrenal zone becomes active in the third trimester, before developing into the aldosterone-producing glomerulosa zone of the postnatal adrenal [10]. Nuchal edema and mild pulmonary hypoplasia were detected at autopsy in our case, but no other hydropic features were noted. The lack of limb contractures and pterygia suggest the oligohydramnios may have been mild and not a long-standing antenatal complication given it was first detected at 21 weeks of gestation. Oligohydramnios, therefore, is unlikely to have significantly contributed to the facial dysmorphism observed in our case.
A specific cause for the growth restriction could not be identified. Although the fetal adrenal has a pivotal role in the regulation of intrauterine homeostasis, fetal development, and maturation (mainly through steroidogenesis) [10], an exact connection between the absent adrenals and growth restriction was not evident. A lack of adrenal glucocorticoids, steroids, and catecholamines is well documented to contribute to impaired mineralocorticoid balance, impaired lung and organ maturation later in gestation, and also influences the timing of labor [10]. These mechanisms are unlikely to be relevant in our case as the infant was delivered in the second trimester, a time when the maternal adrenal and steroid products would have been available to the fetus.
This case reinforces the importance of the perinatal autopsy to verify an antenatal diagnosis, define multiple fetal anomalies or syndromic features, overcome the limitations of ultrasound in the context of oligohydramnios (such as poor visualization), and obtain tissue for histologic, molecular, or biochemical analysis [16]. Importantly, the results of perinatal autopsy can direct genetic counseling, and assist quality assurance and auditing within antenatal ultrasound services to optimize the quality of antenatal imaging. One of the few contexts in which perinatal autopsy may provide less information than antenatal ultrasound is in the setting of significant tissue autolysis, usually related to delayed detection or prolonged fetal death in utero [16].
In conclusion, this report of congenital adrenal agenesis further expands the literature regarding this condition and proves that, although rare, adrenal agenesis does exist as a true pathologic entity. For families with a history of adrenal agenesis, antenatal diagnosis is vital to allow potentially lifesaving treatment to be given as soon as possible after birth in future siblings. In many cases the diagnosis is not detected on antenatal imaging and is confirmed only by accurate autopsy technique. Perinatal autopsy has a critical role to investigate perinatal mortality by providing an opportunity to correlate pathology with antenatal imaging findings and the clinical details. Additional research is required to understand further the pathogenesis of adrenal agenesis.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Dr Virginia Billson for providing expert opinion regarding the postmortem findings and details related to this case, and Associate Professor Duncan MacGregor for assisting with the microscopic imaging.