
Abstract
Cardiac enlargement is a common and important post-mortem finding. The presence of a big heart may provide a basis for determination of the mechanisms of death in an otherwise negative autopsy. In this article we will 1) define terms used to describe the morphology of the heart; 2) describe the ways the heart can enlarge; 3) review the reasons the heart may enlarge and 4) discuss the consequences of that enlargement. In so doing, we hope to assist the pathologist in evaluating cardiomegaly at autopsy and recognizing the significance of the cardiac enlargement.
Introduction
The heart is the seat of the soul. To describe someone as having a “big heart” is to imply one has the abilities of exceptional caring and sympathy. As autopsy pathologists, however, we tend to focus on the heart's other qualities and functions. To us, the heart is a pump - perhaps a sophisticated and important pump - but nevertheless a pump. Its functional component is striated muscle. As such, the heart has a limited way to compensate for a chronic increase in workload or injury, namely to increase the size of the organ. In this article we will 1) define terms used to describe the morphology of the heart; 2) describe the ways the heart can enlarge; 3) review the reasons the heart may enlarge and 4) discuss the consequences of that enlargement. Since cardiac enlargement is a common and important autopsy finding, the hope is that this review will assist the pathologist in evaluating individuals who have enlarged hearts at autopsy and recognizing the significance of the cardiac enlargement.
Definitions
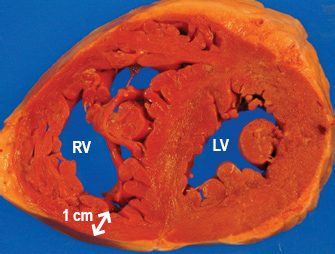
A number of terms are used to describe the size and shape of the heart. Cardiomegaly is a relatively nonspecific term that means “big heart”. Cardiomegaly provides no information regarding which chambers are enlarged, or whether the enlargement is due to hypertrophy, dilatation, or both. Hypertrophy is more specific. Hypertrophy indicates that there is organ enlargement due to enlargement of individual cells. There is no increase in the number of cells. Cardiac hypertrophy can be further described by the shape of the entire organ and its chambers. Eccentric hypertrophy describes a change in the configuration of a hollow organ, such as the heart, in which there is enlargement of the cavities resulting in increased diameter of the organ. Eccentric hypertrophy of the heart is the type that shows up on chest x-ray. On the other hand, concentric hypertrophy, most often used to describe the left ventricle, results in wall thickening without increase in organ or chamber diameter; furthermore, there is a decrease in cavity size. Concentric hypertrophy of the left ventricle does not cause an increase in the cardiac silhouette on chest x-ray. Hypertrophy can involve one or more of the cardiac chambers, but the process is most readily detected in the ventricles first, especially the left ventricle that makes up roughly 75% of the mass of the heart (Image 1).
Atrophy may also affect the myocytes of the heart causing a decrease in mass, usually related to decreased functional demand. Hence, in hearts with severe mitral valve stenosis, the left ventricular mass and dimensions are decreased. Hy-perplasia is an increase in the number of normal cells in an organ. Hyperplasia of the heart is a subject of great interest, as it would be beneficial if myocytes could increase in number after cardiac injury or in response to increased work-load. Unfortunately, if hyperplasia occurs at all, the increase in number of myocytes does not contribute substantially to the mass of the heart after birth (1, 2).

A) concentric hypertrophy in a patient with hypertrophic cardiomyopathy, and B) eccentric hypertrophy with fibrosis (*) in a patient with dilated cardiomyopathy (LV = left ventricle and RV = right ventricle)
Dilation and dilatation are terms used interchangeably to describe enlargement of the cavity of an organ. To a cardiologist, when dilatation is detected clinically by x-ray, ultrasound, or ven-triculography, the expansion of one or more of the cardiac chambers indicates cardiac dysfunction. Dilatation at autopsy is a different matter. If atria appear expanded at autopsy, it is probably a real finding. However, in our opinion, the observation of a dilated ventricle is more problematic. At autopsy, there is no intraventricular pressure against the walls of the ventricles as there is in life. Rigor mortis will contract the ventricles, so a normal heart may actually be more contracted than in end-systole. Autolysis (decomposition), hyperkalemia at the time of death, and acute failure of the heart just before death, may prevent the usual post-mortem contraction of the ventricles. Furthermore, unless the heart is fixed while distended, at physiologic pressures, any post-mortem measurement of ventricular diameter has serious limitations (3).
The last definition to discuss is related to the most catastrophic consequence of cardiomegaly, namely, sudden cardiac death. There are several aspects to the definition of this term. Generally speaking, sudden cardiac death is used when death is “unexpected.” How unexpected does the death have to be? If the individual is known to have underlying heart disease, is an arrhythmic death totally unexpected? How “sudden” is sudden? Some authors say death within one hour of symptoms; others say within 4 hours; others yet say within 24 hours (4). In the absence of clinical electrophysiologic abnormalities and acute cardiovascular abnormalities what is the evidence that the death was cardiac in origin? The role of cardiomegaly in cardiac dysfunction and arrhyth-mogenesis will be discussed in subsequent sections of this review.
Normal Heart Weights and Dimensions
Since the heart has such a limited way to respond to increased pressure or volume load, accurately documenting the degree of hypertrophy or dilatation should provide very useful information regarding the status of the heart. Unfortunately, as discussed, dilatation can not be quantified reliably at autopsy.
Wall thickness is another measure of the degree of hypertrophy. However, wall thickness is dependent on several factors. In concentric hypertrophy, sarcomeres are laid down side-by-side, so wall thickness increases with myocardial mass (5). With chronic dilatation, however, sarco-meres are added end-to-end, so myocardial mass increases while wall thickness may remain the same. Wall thickness at autopsy will also depend upon the degree of contraction of the post-mortem heart. Another problem is that the ventricular wall is highly trabeculated and has papillary muscles. Thus, wall thickness in any given heart is quite variable depending on the site where wall thickness is measured.
Because of all these limitations and because weight is a number that can be measured quite precisely, heart weight is a very useful index of the state of the heart. As luck would have it, the heart weight measurement also has a number of limitations. The heart has a variable amount of epicardial adipose tissue that increases with body weight, steroid use (or production), alcohol use, diabetes mellitus, and other factors. The heart is usually preserved with some segments of great vessels still attached. In addition, the cardiac chambers usually contain post-mortem clots that are often difficult and labor-intensive to remove. Since different chambers may hypertrophy to different degrees, total heart weight does not provide information regarding which chamber has increased the most. In fact, the majority of heart weight is left ventricle, so left ventricular hypertrophy will usually increase heart weight the most. In practice, the one time wall thickness measurements may be more useful than heart weight is in the case of pure right ventricular hypertrophy as occurs in idiopathic pulmonary hypertension, since there will be substantial thickening of the right ventricular wall with little impact on overall heart weight (Image 2). There are methods described to separate the cardiac chambers before weighing. The Fulton method (6) is one, but this method is used primarily for research studies.
Isolated right ventricular hypertrophy from a patient with idiopathic pulmonary arterial hypertension.
How then does one go about obtaining an accurate heart weight? First the heart must be opened before weighing to remove post-mortem clots. The great vessels should be transected 1-2 cm above the semilunar valves. The inferior and superior vena cavae and the pulmonary veins should be cut within 1 cm of their entry into the atria. For routine purposes, epicardial adipose tissue with coronary arteries in place is weighed as part of the heart.
What is normal heart weight? In most mammalian species, including human beings, normal heart weight is 0.45% of body weight for males, and 0.40% of body weight for females, if and only if, body weight is normal (7). Unfortunately, obesity is currently epidemic in industrialized societies, limiting the use of body weight to calculate normal heart weight. For example, in a 400 lb obese individual, 0.45% of body weight would equal 810 gm. This would not be a normal sized heart. In obesity, the heart is enlarged pathologically for a number of reasons. First, adipose tissue is very vascular so cardiac output is increased to supply blood to the fat. Second, in obese individuals, “lean” body mass is increased as muscle is added to carry the excess fat. The increase in skeletal muscle also increases cardiac workload. And third, obesity is associated with diseases that also are associated with hypertrophy of the heart, such as hypertension and diabetes mellitus. Clearly there are serious limitations in using body weight to normalize heart weight. One popular table (8) used by pathologists lists “normal” heart weight for a 5′8″ man as 224-446 gm. This may be the “average” heart weight for this height, but certainly “normal” could not have such a wide range. The articles justifying the use of body weight to normalize heart weight cite a linear relationship between increasing body weight and heart weight. This is precisely the problem. Heart weight does increase with body weight, but this is pathologic; the heart in overweight individuals is not normal.
How then is normal heart weight best estimated? In our opinion, it makes much more sense to normalize heart weight to body height. Zeek, in 1942 (9), did a study of heart size in men and women without heart disease, and came up with formulae based on body height that can readily be used to calculate normal heart weight for adult men and women (Table 1). Table 1 lists information on cardiac dimensions taken from the medical literature, and summarize normal dimensions for adult hearts. Many of these data have serious limitations.
Normal Cardiac Dimensions

Higher than expected, usually normal = ∼.30.
Measured where wall is compact, excluding papillary muscles and trabeculations.
Depends on gender and body size; such wide ranges don't seem physiologic.
In our opinion, the preferred way to calculate normal heart weight in adults.
Works well in lean individuals only.
Causes of Hypertrophy
When the heart is enlarged, regardless of whether it can be designated eccentric or concentric, the hypertrophy is invariably attributed to growth at the cellular level. The heart is an organ with exceptionally low regenerative capacity; the principle mechanism by which the heart responds to increased workload and increased stress is by cardiomyocyte hypertrophy. Protein expression is up-regulated, and sarcomeres, the cell's force-generating machinery, increase in size, resulting in cellular enlargement. Furthermore, the sarcomeres become more organized, arranging themselves in a manner that cumulatively results in either wall thickening (concentric hypertrophy) or chamber dilatation (eccentric hypertrophy). We tend to consider the former a result of pressure overload, and the latter a result of volume overload. However, there are many variables that determine the hypertrophic phenotype (i.e. concentric vs. eccentric).
Before we examine various pathologic entities leading to cardiac hypertrophy (Table 2), let us first consider cardiac enlargement in the absence of disease. Volume overload occurs physiologically in elite endurance athletes as well as pregnant women. This “physiologic hypertrophy” results in an increase in LV chamber size with little or no increase in relative wall thickness (10) —eccentric hypertrophy. “Athlete's heart”, as it is known, is regarded as benign and reversible. However, power athletes, such as professional weight-lifters, undergo a different type of hypertrophy. It is estimated that their cardiomyocytes see transient pressures in excess of 480/350 mmHg, 5-6 days per week, for 5-15 years (11).
Common causes of hypertrophy

The result is pressure overload; their hearts demonstrate concentric hypertrophy. While it is generally accepted that chamber dilatation in endurance athletes is benign, it is well established that, in general, LV wall thickening is damaging to the myocardium, conferring increased risk of arrhythmia and ischemia (12). Moreover, it has been demonstrated that athletes engaging in less extreme strength training, such as rowers, may exhibit LV wall thickening that exceeds the threshold for normal (13). Furthermore, there are data suggesting that LV remodeling in both endurance and power athletes may not completely resolve after long-term deconditioning (14). Thus, the concept of “athlete's heart” is somewhat controversial.
In pregnancy, the heart, as well as other maternal organs, undergoes dramatic structural changes. Maternal cardiac output may increase by 50%, so at parturition the mother's heart is normally heavy. This fact is often overlooked in evaluation of a potential cardiac cause of death of a mother in the peripartum period. “Peripartum” cardiomyopathy can easily be overdiagnosed in this circumstance.
The differential remodeling of pressure overload (concentric hypertrophy) and volume overload (eccentric hypertrophy) is well demonstrated by aortic valve stenosis. In aortic stenosis, the sclerotic, calcified valve causes LV outflow obstruction and increased LV pressure. Structural proteins, such as integrins and stretch receptors, sense pressure overload and initiate a signaling cascade that promotes concentric hypertrophic growth. However, concentric remodeling is not the only pattern of hypertrophy seen in this disease. The increased pressure eventually affects mitral valve function, leading to mitral regurgitation and left atrial volume overload. Left atrial enlargement may be prominent. The LV also becomes volume overloaded, in which case the hypertrophic phenotype is eccentric, not concentric. Furthermore, as the aortic valve becomes more and more sclerotic, it may lose the ability to close completely, resulting in aortic regurgitation. In this case, either concentric or eccentric hypertrophy can be seen (15).
Not all stress to the heart is biomechanical. Neurohumoral factors, such as angiotensin II, catecholamines, and sympathetic innervation, act directly on cardiomyocytes. Neurohumoral and biomechanical stress trigger hypertrophic growth via independent pathways (16). That said, over-stimulation of many these factors also causes hypertension and therefore pressure overload. In such situations, neurohumoral and biomechanical stress act in concert. Though we tend to associate hypertension with concentric hypertrophy—being a disease of pressure overload—studies demonstrate that the geometry of the hypertensive heart is often eccentric (17, 18), perhaps owing to the multifactorial nature of the disease.
Myocardial injury is another important trigger of pathologic hypertrophy—ischemia being a prime example. When injured myocardium is functionally impaired, there is an attempt by surrounding tissue to compensate via eccentric remodeling. The increase in chamber size increases preload, therefore increasing stroke volume and cardiac output. Quite often, however, the compensation is inadequate. As the chamber dilates, wall tension increases (law of Laplace). This increase in stress stimulates further hypertrophic growth, resulting in a downward spiral of maladaptive remodeling. The etiologies of myocardial injury are virtually endless. Important categories to consider include ischemia (coronary artery disease, microvascular disease), myocarditis (infectious, immune-mediated), and toxins (alcohol, chemotherapeutic agents).
Unlike the causes of cardiomegaly previously mentioned, in which external influences trigger cardiomyocyte hypertrophy, dilated and hyper-trophic cardiomyopathies are diseases of the cardiomyocyte itself. It should be noted, that although the term cardiomyopathy is commonly used to describe the hypertrophic heart in general (e.g. ischemic cardiomyopathy), one could argue the term should be reserved for intrinsic diseases of the heart muscle (as it's name denotes). Hypertrophic cardiomyopathy (HCM), previously known as hypertrophic obstructive cardiomyopathy (HOCM), is an inherited cardiomyopathy and an important cause of sudden death in young people (19). It is a disease of the sarcomere, in which defective proteins aberrantly trigger concentric hypertrophy (20). HCM has a peculiar predilection for the interventricular septum, though the entire left ventricle (and occasionally the right) may be affected. Histologically, HCM is characterized by myofibrillar disarray and fibrosis. In contrast to HCM, dilated cardiomyopathy (DCM) represents a diverse group of diseases that share a common phenotype—eccentric hypertrophy. It is estimated that 20-48% of DCM is familial (21), and a wide variety of defective proteins have been implicated. Despite their genomic diversity, the aberrancies all manifest as marked chamber dilatation.
Infiltrative diseases represent a group of rare disorders associated with cardiac hypertrophy. Some are diagnosed early in life, though others may be revealed at autopsy. Grossly the heart may be indistinguishable from cardiomyopathies. However, the histologic appearances are distinct. Infiltrative diseases that may mimic HCM (i.e. concentric hypertrophy) include cardiac amyloid, glycogen storage diseases, primary hyperoxaluria, Fabry's disease, and mucopolysaccharidoses. Examples of infiltrative diseases that mimic DCM (i.e. eccentric hypertrophy) are sarcoidosis, hemochromatosis, and Wegener's granulomatosis (22). Other notable yet uncommon causes of cardiac hypertrophy include muscular dystrophies, neurodegenerative disorders (e.g. Friedreich ataxia), mitochondrial disorders, thiamine deficiency (Beri Beri), and numerous inborn errors of metabolism.
Hypertrophy as a Cause of Death
When is cardiac hypertrophy responsible for death? The hypertrophic heart can result in death in only two ways: 1) failure of the heart as a pump - heart failure, and 2) failure of the electrical system of the heart - arrhythmia or conduction abnormalities. Of course, determination of factors responsible for death involves clinicopathologic correlation - clinical history, laboratory studies, toxicology, etc. If other organ systems have been excluded, then what are the autopsy manifestations of a “cardiac death?”
If death is due to pump failure, then certain anatomic abnormalities should be apparent. If there is left ventricular failure, the lungs should be heavy, and microscopic examination should show diffuse alveolar edema. One caveat is that if a patient dies after failed resuscitation attempts, the lungs are always heavy and full of fluid, so the pathologist can not determine whether or not left ventricular failure was present. The presence of hemosiderin-laden macrophages, so called “heart failure cells” is suggestive but not very specific. If right ventricular failure is present, there should be acute and/or chronic passive congestion of the liver and perhaps peripheral edema.
What if the mechanism of death is arrhythmia or conduction abnormalities? How does a pathologist diagnose an electrical event, if no recordings were made at the time of, or soon after, collapse? Clearly, this is a diagnosis of exclusion. Most sudden cardiac deaths, over 75%, are due to tachyarrhythmias, namely ventricular fibrillation. Less than 25% are due to bradyarrhythmias. The paradigm for the diagnosis of an arrhythmic death is that there are structural abnormalities that predispose to arrhythmias plus a trigger that initiates the arrhythmias (23). Indeed, triggers are often difficult to identify and document; they include physical exertion, emotional stress, fluid/ electrolyte imbalance, hypoxia, and toxins, including pro-arrhythmic drugs.
The structural abnormalities that predispose to arrhythmias are hypertrophy and/or fibrosis. Fibrosis can result in conduction block and re-entry arrhythmias (23). The mechanism by which hypertrophy predisposes to arrhythmia is a complex subject, the discussion of which is beyond the scope of this paper. However, proposed mechanisms include changes in membrane channels, ion pumps, cell receptor abnormalities, and other derangements of the cellular level. Whatever the cellular event, there is no question that hypertrophy of the heart increases the risk of sudden death. The classical Framingham Study (24) found that, in men, hypertrophy of any cause increases the risk of sudden death four fold. Interestingly, in women, hypertrophy only doubles the risk of sudden death. Other studies (25) have confirmed the increased risk of fatal arrhythmia in the presence of cardiac hypertrophy. So finally, having a big heart in life, despite its positive connotations, is likely problematic. However, to the autopsy pathologist, the finding of a big heart is less a problem than a solution. It provides a basis for determination of the mechanisms of death -heart failure or arrhythmia - in what may be an otherwise negative autopsy.