
Abstract
The impacts of chronic high levels of ethanol consumption on cardiovascular structure and function are reviewed as they pertain to forensic pathology practice. Both hypertrophy and remodeling of the myocardium as well as physiological revision of electrical conduction through the heart can occur in a progressive fashion. A discussion about the likely underappreciated spectrum of structural heart disease that may be seen in individuals with chronic alcohol dependence is presented. Finally, brief reference to the collision between alcohol, cardiovascular disease, and forensically relevant scenarios is provided.
Introduction
The purpose of this article is not to extol the virtues of the well-recognized benefits of mild to moderate ethanol consumption in helping to prevent cardiovascular disease, but rather to consider how significant binge drinking as well as heavy, sustained ethanol consumption over years can promote serious cardiovascular dysfunction and in some cases increase the risk of sudden death. The manuscript is written for the forensic pathologist who must frequently consider a history of acute or chronic ethanol intoxication into the assessment of the cardiovascular findings identified at autopsy as well as cause of death determination. This integration need not be simply confined to an evaluation of sudden cardiac death in the setting of a natural manner for death, but may also be extended to non-natural contemplations. Examples could include classical scenarios of trivial assault leading to “homicide by heart attack” in a chronic alcoholic or a case of acute alcohol withdrawal associated with deleterious behavioral consequences that may lead to conflict with others, such as police, only to end with sudden and unexpected death.
It is estimated that 10% of the industrialized world (and a growing percentage of the developing world) are heavy, habitual consumers of ethanol. Literally dozens of pathological processes are either directly or indirectly affected by ethanol consumption; many of which are identifiable at autopsy (
Selected Pathological Changes That May be Identified at Autopsy Related to Chronic Heavy Alcohol Consumption *
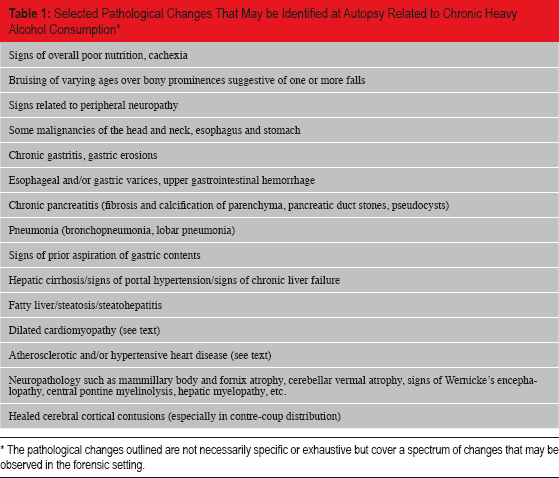
The pathological changes outlined are not necessarily specific or exhaustive but cover a spectrum of changes that may be observed in the forensic setting.
Whereas moderate consumption (three to nine drinks per day) has been associated with decreased atherosclerotic coronary artery disease, stroke and all cause mortality (1), excess chronic consumption is known to have adverse effects. It must be recognized that most studies considering a dose-response relationship between alcohol and disease were performed largely when considering the pathophysiological consequences for the living, with little clear understanding available about the cumulative burden of alcohol induced cardiac pathology identified at autopsy, for those who were either symptomatic or asymptomatic in life. Moreover, if we accept that there may be a three- to four-fold variation in the rate of alcohol metabolism between individuals (2), an imperfect causal relationship arises between the quantity of ethanol consumed and the presence of underlying structural pathology of the heart. Furthermore, because of the wide spectrum of alcohol-associated cardiac findings identified, their nonspecific nature, and the inherent variability with which individuals develop structural cardiac pathology (i.e., not all heavy drinkers develop dilated cardiomyopathy), it is likely that alcohol-associated cardiovascular disease is underrecognized at autopsy, underscoring the difficulty in ascribing an accurate incidence in the population.
Chronic ethanol consumption is thought to cause damage to cardiomyocytes through multiple possible pathways. Not only is ethanol itself directly toxic, but its metabolites (in particular, acetaldehyde) can exert direct toxic effects. These effects can include altered gene expression, dysfunction, or damage to structural and sarcomeric proteins in heart muscle cells, mitochondrial dysfunction, alterations in calcium homeostasis, and changes in cell surface receptors and signal transduction pathways (3). These changes can impair excitation-contraction coupling, oxidative phosphorylation for energy metabolism, and contractility. More global modifications to regulation of the renin-angiotensin-aldosterone system and lipoprotein metabolism are also recognized that can have significant secondary effects on the heart and vasculature (1). Other secondary pathologies to consider with chronic ethanol consumption that may impact the heart could include malnutrition with vitamin deficiency, especially thiamine, as well as mineral and electrolyte deficiencies that include selenium, phosphorus, potassium, and magnesium (1). Malnutrition-associated ketosis can also have a more acute impact on myocardial performance. Finally, the historic inclusion of lead or cobalt in alcoholic beverages may lead to direct cardiotoxic effects.
Structural Heart Disease and Chronic Ethanol Consumption
There is a broad spectrum of macroscopic and microscopic changes that may be observed in the setting of ethanol induced structural disease of the heart. However, these changes can perhaps be categorized into two principal (and not mutually exclusive) groups to facilitate thinking about the underlying pathophysiology and structural changes: 1) the classical dilated cardiomyopathic phenotype and 2) changes supportive of atherosclerotic and hypertensive heart disease.
Alcohol-Associated Dilated Cardiomyopathy
The classical cardiac pathology envisioned when considering heart muscle disease and chronic ethanol dependence is that of a dilated cardiomyopathy (DCM). Dilated cardiomyopathy generally presents at a relatively young age, often during the fifth decade of life, and typically requires many years of chronic, sustained heavy consumption of ethanol. It is estimated that the prevalence of cardiomyopathy amongst alcoholics is variable and ranges from 23%-40%, more frequently in men than it is in women (3). Most men who develop alcohol related DCM have consumed more than 80 g of ethanol per day over a long time period, which roughly correlates with one liter of wine, eight standard beers, or half a pint of liquor (1). Interestingly, there may appear to be an inverse correlation between the presence of cirrhosis and DCM in chronic alcoholics, where frequently if DCM is present, cirrhosis may not be (4). The precise underlying etiology for DCM is not well understood, but likely involves many of the mechanisms discussed above.
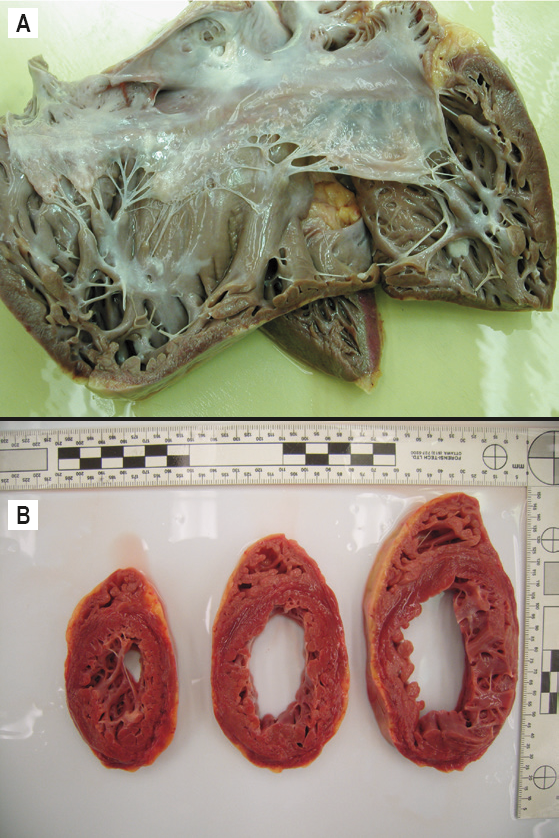
This secondary cardiomyopathic process (a “toxic” cardiomyopathy) is nonspecific in its macroscopic appearance (
The microscopic findings observed in alcohol-associated dilated cardiomyopathy are also nonspecific in their appearance (
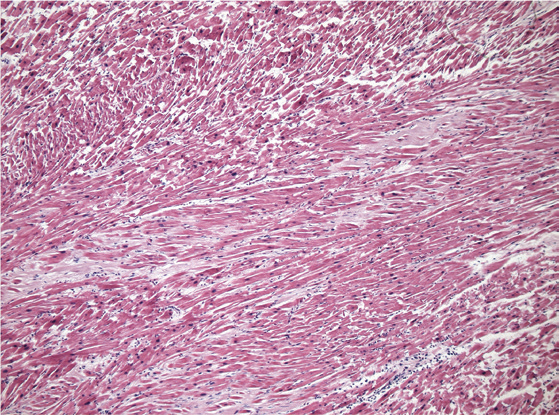
Microscopic image of the myocardium in a case dilated cardiomyopathy as can be seen in chronic ethanol usage. The histological findings are nonspecific but generally include cardiomyocyte hypertrophy, interstitial and perivascular fibrous tissue deposition, and cardiomyocyte degenerative changes (H&E, x200).
Alcohol-Associated Atherosclerotic and Hypertensive Heart Disease
It is not as well recognized that chronic, sustained, heavy ethanol consumption increases the risk of developing atherosclerotic coronary artery disease and systemic arterial hypertension. These pathological substrates can also lead to remodeling and hypertrophy of the heart, increase the risk of developing ventricular arrhythmias and consequently, sudden cardiac death. Moreover, heavy chronic consumption of alcohol may act to potentiate the pathophysiological effects of classical risk factors that also promote ischemic heart disease.
When chronically ingested in significant quantities, ethanol can promote ventricular systolic and diastolic dysfunction (heart failure with reduced ejection fraction and heart failure with preserved ejection fraction, respectively), systemic arterial hypertension, angina pectoris, ventricular arrhythmias, and sudden cardiac death (1). Increased stiffening and hypertrophy of the left ventricle follows not only from the direct toxic effects of ethanol on the myocardium with loss of cardiomyocytes and interstitial fibrous tissue deposition, but also from stimulation of systemic arterial hypertension, which promotes left ventricular hypertrophy and development of hypertensive heart disease. It is estimated that chronic ethanol consumption is a causative factor for just over 10% of cases of hypertension. This influence is prominent when ethanol consumption exceeds 30 g per day and may reflect an increase in sympathetic outflow as well as plasma levels of catecholamines, renin, and aldosterone (1,6). However, like the propensity to develop DCM, the dose-response relationship with ethanol in such circumstances is imperfect.
Heavy alcohol consumption inhibits oxidation of free fatty acids and stimulates triglyceride synthesis. This can result in elevated levels of very low-density lipoproteins (VLDL), low-density lipoproteins (LDL), and total cholesterol. In addition to hypertension, such changes may promote atherogenesis (1). This is in contrast with elevation of high-density lipoproteins (HDL) with more moderate consumption of ethanol and a reduced risk of atherosclerosis. Because of the increasing prevalence of traditional risk factors for atherogenesis and hypertension in our society, it is difficult to estimate the contribution played by chronic ethanol consumption in those with ethanol dependence.
Functional Heart Disease and Ethanol Consumption
Acute ethanol intoxication, when significant, can increase the risk of arrhythmia. Ethanol has been associated with a number of atrial and ventricular dysrhythmias that include atrial and ventricular premature beats, supraventricular tachycardia, atrial flutter and fibrillation, and ventricular tachycardia and fibrillation (1). It is typically the rapid ventricular arrhythmias that may be associated with unexpected death.
Arrhythmogenesis Associated with Acute and Chronic Alcohol Consumption
Atrial fibrillation, manifesting as “holiday heart,” is the arrhythmia most commonly associated with acute ethanol intoxication, although multiple factors should also be considered. First, the timing of the acute intoxication is not important per se, (i.e., it is not required to occur on a holiday), although there does appear to be a correlation with dosage as higher blood alcohol concentrations increase the risk and most episodes appear to occur after binge drinking. Demographic factors such as age and concurrent pathological factors such as aortic and mitral valvular disease, systemic arterial hypertension, and left ventricular dysfunction may also increase the risk of atrial fibrillation. This is likely due in part to the propensity of these conditions to increase the size of the left atrial chamber as the result of chronic pressure and volume changes.
Furthermore, a chronic high level of ethanol consumption by itself has also been associated with an increased risk of atrial fibrillation. This may be due to the pathophysiological consequences of structural disease of the ventricular myocardium as well as the direct toxicity to the atrial cardiomyocytes themselves. Clinical studies have not identified a robust correlation between moderate ethanol consumption and atrial fibrillation, but only when alcohol is imbibed in larger quantities (7).
Prolongation of the QTc interval is recognized in chronic alcoholics who are not acutely intoxicated, which is supportive of underlying structural and likely molecular alterations to the myocardium. Acute ethanol intoxication at moderate or higher levels is also associated with prolongation of ventricular polarization (8). This effect was observed regardless of the presence of atherosclerotic coronary artery disease. The P wave is also prolonged in moderate to severe, acute ethanol intoxication (9). The significance of this observation is supported by anecdotal evidence of third degree atrioventricular blockade following acute ethanol intoxication (10,11).
Evaluating the role of subclinical myocardial damage in a habitual “social drinker” that predisposes to arrhythmia versus developing an arrhythmia due to acute intoxication alone is difficult to evaluate in most clinical studies. There is little evidence to say what minimum exposure to alcohol is necessary to develop myocardial pathology that can be identified at autopsy. Does one need to be classified as an alcoholic to potentially possess subclinical disease? The answer is likely no. The absence of a history of congestive heart failure per se does not negate the risk of sudden and unexpected death if sufficient end organ damage is identified in an appropriate context. It might be suggested that the forensic pathologist sees a greater spectrum of end organ damage in the form of cardiomyocyte hypertrophy and fibrous tissue deposition from chronic ethanol consumption than the clinician who must rely on clinical presentation as well as the limitations of electrophysiological and echocardiographic modalities. Nevertheless, multiple case reports have suggested that severe acute intoxication alone in a relatively naïve user can increase the risk of ventricular tachyarrhythmias—a risk that appears to subside days after the intoxication (12).
Myocardial remodeling (from any cause) that is associated with interstitial fibrous tissue deposition, ventricular hypertrophy, and autonomic dysfunction is recognized to increase the risk of ventricular dysrhythmia. When the structural changes are severe enough, particularly when cardiomyocyte degenerative changes are noted (although this often requires extensive sampling of the heart), a diagnosis of cardiomyopathy may be reasonably applied. It is difficult to discern the relative pathophysiological contributions of acute from chronic alcohol consumption when considering a sudden cardiac death in a chronic alcoholic. It may be the case that the risk of ventricular arrhythmia in severe acute ethanol intoxication in a chronic alcoholic is at least additive if not synergistic with the risk posed by the underlying structural heart disease itself. However, although such a conclusion is reasonable based on the coincidence of arrhythmogenic mechanisms present, this premise is ultimately speculative as it is difficult to weigh the relative contributions of each factor clinically or at autopsy. Finally, sudden and unexpected death in a chronic alcoholic with little or no ethanol identified in the postmortem blood, a fatty liver with or without cirrhosis and little else is a well-recognized forensic pathological issue. This may represent a sudden arrhythmic death due to fluid and electrolyte abnormalities, sudden arrhythmic death due to structural disease in the heart (or both), or possibly sudden death in the setting of acute ethanol withdrawal or from alcoholic ketoacidosis. Certainly, other metabolic considerations could also be involved. When the alcoholic is found deceased with no supportive historical information available to assist with the death investigation, multiple such considerations should be evaluated.
Arrhythmia and Acute Ethanol Withdrawal
Acute ethanol withdrawal is a syndrome that can occur in individuals chronically and physiologically dependent on ethanol who sustain a significant decrease or complete cessation of ethanol consumption. The spectrum of signs and symptoms in severe cases can include autonomic hyperactivity, tremulousness, diaphoresis, nausea/vomiting, hypertension, tachycardia, hyperthermia, and tachypnea. More ominous symptoms in severe cases include withdrawal hallucinations, withdrawal seizures, and delirium tremens (13). Delirium tremens combines autonomic hyperactivity with other symptoms such as disorientation, confusion, delirium, psychosis and seizures. While the symptoms of alcohol withdrawal syndrome can begin to be observed two to six hours after a decrease in ethanol ingestion, severe manifestations such as delirium tremens often do not develop for many more hours (48–72 hours) after a decrease in ethanol consumption. Prior to supportive treatments such as hydration, fever control, nutrition, and benzodiazepine treatment, the mortality rate was as high as 35% in those with delirium tremens (13).
There is anecdotal evidence of an increased risk of ventricular arrhythmia in humans as well as in animal studies in the setting of ethanol withdrawal syndrome (14,15). The precise mechanism for this is not clear; however the autonomic instability, adrenergic hyperactivity as well as frequent fluid and electrolyte abnormalities observed in chronic alcoholics with acute withdrawal are likely contributing factors. Furthermore, the aforementioned structural heart disease that may be found in those with a history of chronic, heavy ethanol consumption can provide an additional pathological substrate for arrhythmogenesis that may act in conjunction with the pathophysiological derangements observed with alcohol withdrawal. It must also be remembered that acute metabolic derangements such as alcoholic ketoacidosis may also cause sudden and unexpected death and must be considered in relevant cases. Finally, it is in the setting of ethanol withdrawal syndrome with agitation and/or psychosis that a possible confrontation with health care workers, security guards, or police can result. Although uncommon, significant confrontations have been followed by sudden and unexpected death, particularly if a struggle has ensued. Under such circumstances, the pathophysiological consequences of chronic ethanol intoxication and acute ethanol withdrawal on the heart become quite relevant and must be balanced against other factors in the investigation.
Cocaethylene
A few words should be said about the metabolite, cocaethylene, and its apparently increased cardiotoxicity. Cocaethylene is a covalently modified, active metabolite of cocaine formed in the presence of ethanol during the antemortem period. Multiple lines of evidence have suggested that cocaethylene exhibits a greater lethality than cocaine alone, which is thought to occur through an arrhythmic mechanism. Cocaethylene is recognized to have a longer elimination half-life than cocaine, which may potentiate its negative pathophysiological consequences for the heart (14).
Conclusion
When assessing the potential role of ethanol consumption in contributing to cardiac pathology or sudden cardiac death, it is important to consider the full history available in the context of the scene and a full postmortem examination. Acute and/or chronic ethanol consumption may contribute directly or indirectly to death, and may do so by causing structural and/or functional disease. As a practical point, quantitatively estimating the effects of ethanol on the heart in the context of other environmental, toxic, and genetic factors that may contribute to heart disease is virtually impossible at autopsy. Most middle-aged and older individuals that are autopsied exhibit some degree of heart disease with a history that is often supportive of multiple risk factors. Thus, when the history of ethanol consumption is known to be high (whether acutely or chronically), then including ethanol as a significant factor to consider when evaluating sudden death in an individual with heart disease may be reasonable, but it must be considered in the context of the entire case and corroborate other forensically relevant opinions borne from the death investigation.
Footnotes
The author, reviewers, editors, and publication staff do not report any relevant conflicts of interest.