
Abstract
In forensic pathology, genetic testing is a luxury that is not readily entertained. With molecular advances, genetic testing can provide critical information to conclude cause and manner of death in otherwise sudden, undetermined deaths. An autopsy was performed on a 30-year-old woman who died suddenly three months postpartum. The patient's history was unremarkable and included a negative electrocardiogram test prior to pregnancy. Toxicology results were negative and there were no gross or histologic findings on a complete postmortem examination including the conduction system. Prompted by personal interest and knowledge, the spouse, an electrophysiology fellow, requested genetic testing be performed and personally financed the tests. The results revealed two genetic mutations strongly associated with Type 1 Long QT Syndrome, expected to be familial. This provided a cause and manner of death and also instigated preventive measures for testing in the child. This case supports previous studies that acknowledge the need for molecular testing in the autopsy-negative, sudden unexpected death cases in young patients. Forensic pathologists have a duty to the public to serve as their physicians and this may prove to be a key area that can contribute to the preventative medicine effort and provide closure to families that may have loved ones left with an undetermined cause and manner of death. Although financially a problem, the future of pathology is undeniably heading in the direction of molecular testing and an attempt to adjust an internal budget in order to include expenses for these tests should be considered.
Introduction
In the forensic setting, sudden unexpected death (SUD) requires investigation to yield a cause and manner of death. Although most of these cases are natural in manner, determination of the cause at times can be problematic when a lack of gross or histologic evidence is encountered. The overwhelming incidence of sudden cardiac death within the United States, reported as high as 400 000 deaths per year, is many times accompanied by gross or histologic findings consistent with a cardiovascular origin (1). However, for cases of SUD especially in the young, the possibility of a genetic mutation inducing a fatal arrhythmia should be investigated when autopsy fails to reveal any gross or histologic cardiovascular abnormalities. As molecular testing begins to become standard practice in pathology, the term “molecular autopsy” has emerged to encompass postmortem genetic testing in an otherwise negative autopsy. With the ability to test for a variety of mutations, cardiac channelopathies have proven to be significant pathogenic disorders causing sudden unexpected death in the young.
One of the more significant clinical syndromes that carries detectable genetic mutations and can induce sudden cardiac death is long QT syndrome (TQTS). A variety of mutations associated with this syndrome impair cardiac channels so that the ability of myocardial repolarization is delayed, causing a prolongation of the QT interval on electrocardiogram (ECG) and predisposing the individual to torsades de pointes and subsequent sudden cardiac death (2). Autosomal dominant and autosomal recessive forms of inheritance have been identified, raising the question as to what role pathologists play in preventative medicine and the duty and resources available to work with when counseling the families potentially affected. In addition to inheritable mutations, the possibility of forming acquired LQTS has also been identified (3). Occasionally, these individuals may be silent carriers and may only require an arrhythmogenic trigger such as drugs, physical exertion, or emotional stress to induce a fatal arrhythmia (2). Although many patients with LQTS will have clinical evidence of their disease including previous episodes of syncope, bradycardia or seizure, the actual prolongation of the QT interval may not be identified on a baseline ECG (2). For those patients without previous clinical signs or symptoms that experience SUD, the ability to detect this syndrome and reliably identify a cause and manner of death should be available and pursued.
Case Report
The decedent was a 30-year-old woman with a past medical history of mild asthma and a cesarean delivery three months earlier. Prior to maternity leave, she was employed as a clinical pharmacist. She lived at home with her husband and 3-month-old daughter. She had no recent complaints. On the morning of her death, her husband went to work and she went to shower. After not emerging from the bathroom, a family member staying at the home entered the bathroom to find her unresponsive on the floor. She was transported by emergency medical services to a nearby hospital where she arrived in full arrest and was pronounced dead approximately ten minutes after arrival.
At the time of autopsy, the body was that of a normally developed, adequately nourished, atraumatic female. A complete postmortem examination showed evidence of therapeutic intervention with pulmonary congestion and edema, but failed to reveal an anatomic cause of death and a pending death certificate was issued. Samples were taken including femoral blood that was transferred to EDTA tubes and frozen for potential future testing. Review of histological sections, including the cardiac conduction system, failed to disclose her cause of death.
Initial investigation of the scene did not suggest any foul play. The investigating agency later requested further toxicologic testing, prompted by concerned parties, which yielded negative results.
At the time of her death, the decedent's husband was employed as an electrophysiology fellow, and upon receiving the initial results of the autopsy, decided to pursue and personally finance genetic testing. Of note, the husband, in preparation for pregnancy, had previously performed an ECG on the decedent that had not revealed any abnormalities. At his request, the femoral blood collected at the time of autopsy was sent for genetic testing, which consisted of a panel of cardiac channelopathies including those mutations associated with LQT1 through LQT6. The results of this test revealed two probable deleterious mutations involving the KCNQ1 gene, associated with Long QT Syndrome Type 1. Each of these mutations was a point mutation that impacted the alpha subunit of the voltage-gated potassium channel at a separate location
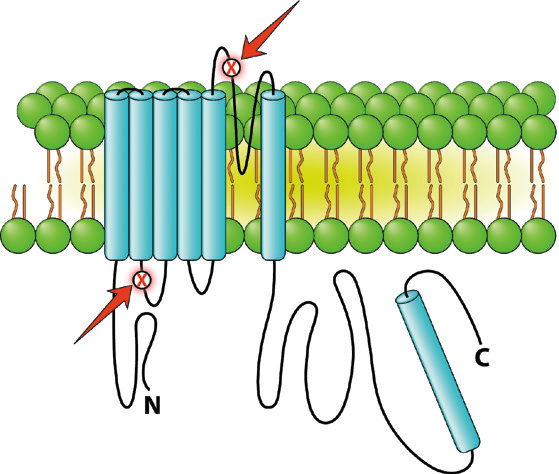
Depiction of the two KCNQ1 mutations (red arrows) identified in the decedent encoding the alpha subunit of the cardiac ion potassium channel associated with Long QT Syndrome, Type 1 (LQT1). Image created by: Patrick Mooney Biomedical Illustrator/Animator.
Discussion
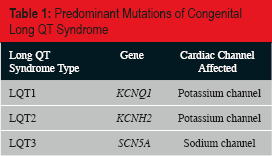
Suspicion of an underlying genetic abnormality should be heightened in any case of sudden unexpected death with emphasis on those less than 40 years old in which the likeliness of underlying natural disease not genetic in origin is considerably less. Long QT syndrome is a complex syndrome with hundreds of mutations identified. Most of these mutations have been localized to a handful of genes that allow classification of this syndrome into long QT types 1 through 12 (LQT1–12) (3). Many of these mutations result in abnormalities of the potassium channel including LQT1 (40–55%) and LQT2 (35–45%), caused by mutations in genes KCNQ1 and KCNH2 respectively, resulting in a delayed action potential and an increase in the QT interval (4, 5). Additional cardiac channels affected by various gene mutations include sodium and calcium, with occasional membrane proteins being affected (2). The majority (>90%) of congenital LQTS are attributed to mutations resulting in LQT1 through LQT3
Predominant Mutations of Congenital Long QT Syndrome
The inheritance of these genetic mutations can be either autosomal dominant, resulting from any of the mutations currently identified, or as autosomal recessive, which is associated with LQT1 and LQT5 and presents with sensorineural hearing loss in addition to cardiac abnormalities (2). The penetrance of these mutations can be low, resulting in individuals that are silent carriers (3). These individuals, as well as their children, are at increased risk of SUD if exposed to arrhythmogenic triggers including drugs or metabolic disturbances (2, 3). In a study of nine cases of LQTS, 15 of 46 family members who tested positive for genetic mutations were silent carriers (33%) (6). In addition, sporadic mutations have been identified and may be associated with sudden infant death syndrome (SIDS) (7).
Different types of LQTS have been shown to be associated with certain demographic and clinical presentations. This can be an important tool for forensic pathologists when assessing the need for genetic testing. For instance, the incidence of certain mutations has been documented by age and presentation with a decrease in LQT3 in those patients experiencing cardiac events before the age of ten compared to LQT1 and LQT2 (8). LQT1 has also been shown to be more common in patients experiencing cardiac events during exercise as compared to those at rest with LQT3 and emotional stress in LQT2 (4, 9). Most importantly from a forensic standpoint, lethal cardiac events have been shown to be most common in patients with LQT3 (2).
In addition to those mentioned, postpartum patients have been shown to have an increased risk of precipitation of arrhythmias if harboring a long QT mutation (10). During pregnancy, cardiac output increases to compensate for the fetus and the increase in blood volume, which results in an increased heart rate. For those individuals with mutations, this results in a shortened QT interval (10). In the postpartum period, the heart rate and cardiac output decreases, which may unmask a prolonged QT interval resulting in cardiac events, especially when compounded by the physiological stress of becoming a new mother. In one retrospective study from the University of Rochester, 111 individuals harboring long QT mutations were analyzed to reveal that these individuals were more likely to have cardiac events during the postpartum interval (23.4%) versus the pre-pregnancy period (3.8%) (11). In a separate study that looked at 388 women who underwent genetic testing, postpartum cardiac events were more commonly associated with LQT2 (16%) than LQT1 (<1%) (12). Although the case presented in this report consisted of mutations associated with LQT1, this highlights the possibility of LQTS being present in postpartum patients experiencing SUD.
It is known that the penetrance of this syndrome is variable; therefore, a thorough clinical history including a family history should be obtained whenever possible. In one of the largest studies performed to date, the Mayo Clinic looked at 173 cases of SUD in which a “molecular autopsy” was performed. Although this study included catecholaminergic polymorphic ventricular tachycardia (CVPT) mutation detection in addition to LQTS mutations, 45 cases (26%) were positive for at least one genetic mutation and with thorough investigation, the medical examiner was able to document a family history of sudden cardiac death in 20 cases (11.6%) (1). Clinical history is imperative in cases of SUD; however, the evidence of a previously negative antemortem ECG cannot be used to rule out LQTS. Although the name implies a prolongation of the QT interval, studies have shown that antemortem ECGs may be negative in patients harboring long QT mutations (13, 14). These findings, in combination with the case report presented here, underscore the need for thorough investigation and correlation.
We know that genetic mutations can be present in cases encountered in the forensic setting of sudden unexpected death, especially in the young or in certain situations such as the case presented. The difficulty then becomes the diagnosis or detection of these mutations when clinical history is limited and the ability to finance such testing is not available. Many families cannot afford to personally finance genetic testing, which can be as high as $2500 per panel, as was done in our case. The burden then falls on the medical examiner's office and the forensic pathologist when a genetic mutation is suspected. Unfortunately, there are limited studies to illustrate the cost-effectiveness of such testing (15).
Prior to the Mayo study, only occasional smaller studies and case series were available for data extraction, which revealed the presence of LQTS mutations in 7–38% of cases of SUD
Proportions of Long QT Mutation-Positive Cases in Previously Reported Studies Which Performed Genetic Testing

Exclusion criteria in this sample (n=270) was age ≤ 20 years old.
Attempts have been made to provide an algorithmic approach to these cases in a clinical setting as well as in the postmortem setting that may help curb the cost accrued by these cases and should be taken into consideration when faced with a SUD. A Schwartz score has been developed for the diagnosis of congenital LQTS that takes into account ECG findings, clinical findings, and family history, which may be a useful tool for forensic pathologists when thorough medical records can be obtained (13). A tiered approach was also created based on data extracted from a study using 430 known LQTS patients (21). These recommendations began with suspected LQTS patients undergoing genotyping for a set of frequently mutated codons, yielding diagnoses in 40–50% of cases. A negative result would prompt further screening of the KCNQ1 and KCNH2 coding regions (21). Although a clinical study, this algorithm approach can be applied in the forensic setting.
As mentioned, in order to be cost effective and ensure results that will be useful, testing should be carried out in a targeted manner. As with any toxicology tests, for example, a panel of testing includes those that will yield the most information, many times with the aid of a toxicologist to help with interpretation of atypical results. Fortunately, companies that offer genetic testing, such as in our case, also provide information to help interpretation within the report. This may include classifying the mutations detected to help the ordering physician decide whether or not the results are significant. For the forensic pathologist, this can help with determining the difference between a noncontributing positive test result and a result that can be used to determine the cause of death in conjunction with the history. For instance, the company that provided testing in our patient categorized the mutations into classes based on how many unrelated probands with the syndrome carry the mutation as well as the absence of the mutation in large control populations based on published literature. Of course, regardless of the test results, the forensic pathologist will use his or her experience, judgment, and the circumstantial evidence to determine the likely cause of death rather than use a single lab test as a diagnosis to render the cause.
Conclusion
In a sense, the forensic pathologist serves as a physician to the general public when determining the cause and manner of death of individuals that may impact others. Being aware of decedent's clinical history and presentation with thorough investigation is the first step in identifying SUD in patients with possible LQTS. Genetic analysis not only provides information for the forensic pathologist to reliably conclude a cause and manner of death and provide closure for the family; it may lead to additional genetic testing in family members or utilization of genetic counselors for prevention of future SUD. Fortunately, there are also organizations that focus on this problem - most notably the “SADS (Sudden Arrhythmia Death Syndromes) Foundation” - that can serve as invaluable tools to the forensic pathologist to better understand and address these cases. Forensic pathologists have a duty to the public to serve as their physicians and this may prove to be a key area in which they can contribute to the preventative medicine effort and provide closure to families that may have loved ones left with an undetermined cause and manner of death.
As molecular tests continue to become technically advanced and the availability of the “molecular autopsy” is brought to light, consideration should be given to funding of these tests. Although adjusting an internal budget may be problematic or even impossible in some offices, at minimum, an effort to inform the family of the potential genetic consequences should be provided, allowing for the option of personally financed testing if desired. As the future of pathology continues to evolve, the “molecular autopsy” may provide the ability for forensic pathologists to render cause and manner of death when faced with diagnostic difficulty and to better serve the public as physicians.
Footnotes
The authors, reviewers, editors, and publication staff do not report any relevant conflicts of interest.