
Abstract
The E4 allele of apolipoprotein (apoE4) is the primary genetic risk factor for late onset Alzheimer’s disease (AD), yet the exact manner in which apoE4 leads to the development of AD is undetermined. Human and animal studies report that apoE4-related memory deficits appear earlier than the AD clinical manifestation, thus suggesting the existence of early, pre-pathological, apoE4 impairments that may later lead to AD onset. While current research regards the hippocampus as the initial and primary effected locus by apoE4, we presently investigate the possibility that apoE4 innately impairs any brain area that requires synaptic plasticity. To test this hypothesis, we trained young (3-4-month-old) target-replacement apoE3 and apoE4 mice in conditioned taste aversion (CTA) acquisition and extinction learnings— hippocampus-independent learnings that are easily performed at a young age. Synaptic vesicular markers analysis was conducted in the gustatory cortex (GC), basolateral amygdala (BLA), medial prefrontal cortex (mPFC), and hippocampal CA3 to reveal underlying apoE4-related impairments. We have found that young apoE4 mice are severely impaired in CTA acquisition and extinction learning. CTA acquisition impairments were correlated with reduced vGat and vGlut levels in the BLA and GC, but not in the CA3. CTA extinction was correlated with lower synaptophysin and vGlut levels in the mPFC, a central region in CTA extinction. Our results support apoE4-related early-life plasticity impairments that precede the AD clinical manifestations and affect any brain area that depends on extensive plasticity; early impairments that may promote the development of AD pathologies later in life.
Keywords
INTRODUCTION
Several genetic risk factors are correlated with late-onset Alzheimer’s disease (AD), with the ɛ4 allele of apolipoprotein E (apoE4) being the most prevalent one [1, 2]. The other two major alleles, apoE3 and apoE2, are considered to be neutral and protective against AD, respectively [1]. In the brain apolipoprotein E is mainly produced by astrocytes and serves as a cholesterol and lipids transporter [2]. The relation between apoE4 and AD is not well understood. ApoE4 abnormalities have been mostly linked with old-age hippocampal-related memory impairments corresponding to the late-onset AD pathologies found predominantly in the hippocampus. These pathologies include elevated amyloid-β plaques and neurofibrillary tangles [3, 4] in addition to progressive neuronal loss [5]. However, less is known about the basal abnormalities of apoE4 in intact brain tissues and their impact on different memory systems. This information is highly valuable since it may provide an insight into the initial, pre-pathological, effects of apoE4 that may later lead to the development of AD pathologies. It may also explain early human learning deficits which precede AD onset [6–8], and their corresponding animal studies showing early structural synaptic abnormalities [9–11] and hippocampal learning and memory alterations [11–13].
ApoE4 is expressed throughout the brain and was shown to affect neuronal plasticity [14–17]. We hypothesize that even though these effects were described in the hippocampus, and hippocampal related pathways [14–17], the impact of apoE4 should be diffused and evident in any neuronal system undergoing plasticity. Specifically, in this study we focused on the extra-hippocampal conditioned taste aversion (CTA) acquisition and extinction paradigms in young target replacement humanized apoE4 and apoE3 mice. CTA is a robust classical conditioning learning procedure [18, 19] in which a novel palatable taste becomes aversive following association with experimentally-induced gastrointestinal malaise. Post-CTA exposures to the taste alone promote extinction learning. Importantly, extinction learning is not an erasure of a previously acquired memory and it involves extensive synaptic plasticity similar to CTA acquisition [20, 21].
CTA possesses several advantages in the context of the current study. First, it does not require an intact hippocampus [22–24] but involves synaptic modulations primarily in the gustatory cortex (GC) [25, 26], the basolateral amygdala (BLA) [27, 28], and the medial prefrontal cortex (mPFC) [29, 30]. Since these areas are largely intact in young apoE4 mice [10, 31], they may assist in revealing the manner in which apoE4 affects normal brain functioning. Second, task complexity can be finely manipulated using different concentrations of the taste and/or the malaise-inducing agent and thereby allow us to avoid either a floor or a ceiling behavioral effect [32]. Lastly, CTA is based on naturally innate rodent behavior and therefore is highly robust, fast (single exposure) and easily achieved in young animals. Therefore, CTA procedures may serve as a unique tool for researching early apoE4 anomalies.
In this study we have demonstrated that young apoE4 mice are impaired in CTA acquisition and extinction. These behavioral impairments are associated with reduced structural plasticity in the extra-hippocampal brain areas that support these learnings, as compared to apoE3 mice. Thus, our findings support the existence of an early task-dependent and brain-region independent apoE4-related plasticity impairments.
METHODS
Mice
Target replacement apoE4 female and male mice were purchased from Taconic Laboratories (Germantown, NY) and were prepared as previously reported [33]. To minimize possible genetic drift between the apoE4 and apoE3 mice, they were further crossed by us with wild type C57BL/J6Rcc-HSD mice. Mice were housed in cages of similar gender on a 12 h light/dark schedule until the experiment when they were 3-4 months old. Mice were given ad libitum access to chow and water, unless otherwise specified. All methods complied with the Tel-Aviv University Institutional Animal Care Ethics Use Committee guidelines.
Experimental design
Conditioned taste aversion (CTA) acquisition and extinction
The behavior procedures are illustrated in Fig. 1A and B. Mice were transferred to individual cages 12 h before habituation to the experimental setting. During the habituation trials (Fig. 1A[i-ii]), water was provided for 20 min using two 10 ml surgical pipettes once in 12 h until the total water consumption surpassed 1 ml in two consecutive trials (usually taking 4–7 trials). On the following conditioning trial (Fig. 1A[iii]), the same procedure was repeated except that 0.2 M sucrose solution replaced the water in the spouts. Ten min after trial termination, mice were given 0.15 M lithium chloride (LiCl) intraperitoneal injection at either 1% (weak conditioning) or 2% (strong conditioning) of body weight to induce gastrointestinal malaise. During the subsequent trial (Fig. 1A[iv]), water was given again in the two pipettes. A two-pipette test was conducted 24 h after conditioning: Two randomly positioned pipettes were inserted into each cage simultaneously, one containing water and the other 0.2 M sucrose solution (Fig. 1A[v]). Sucrose preference was calculated as: [Sucrose intake (ml)]/[Sucrose intake(ml)+water intake (ml)]. To determine the degree of CTA extinction after strong conditioning, the two-pipette test was repeated for 7 consecutive trials (Fig. 1B).
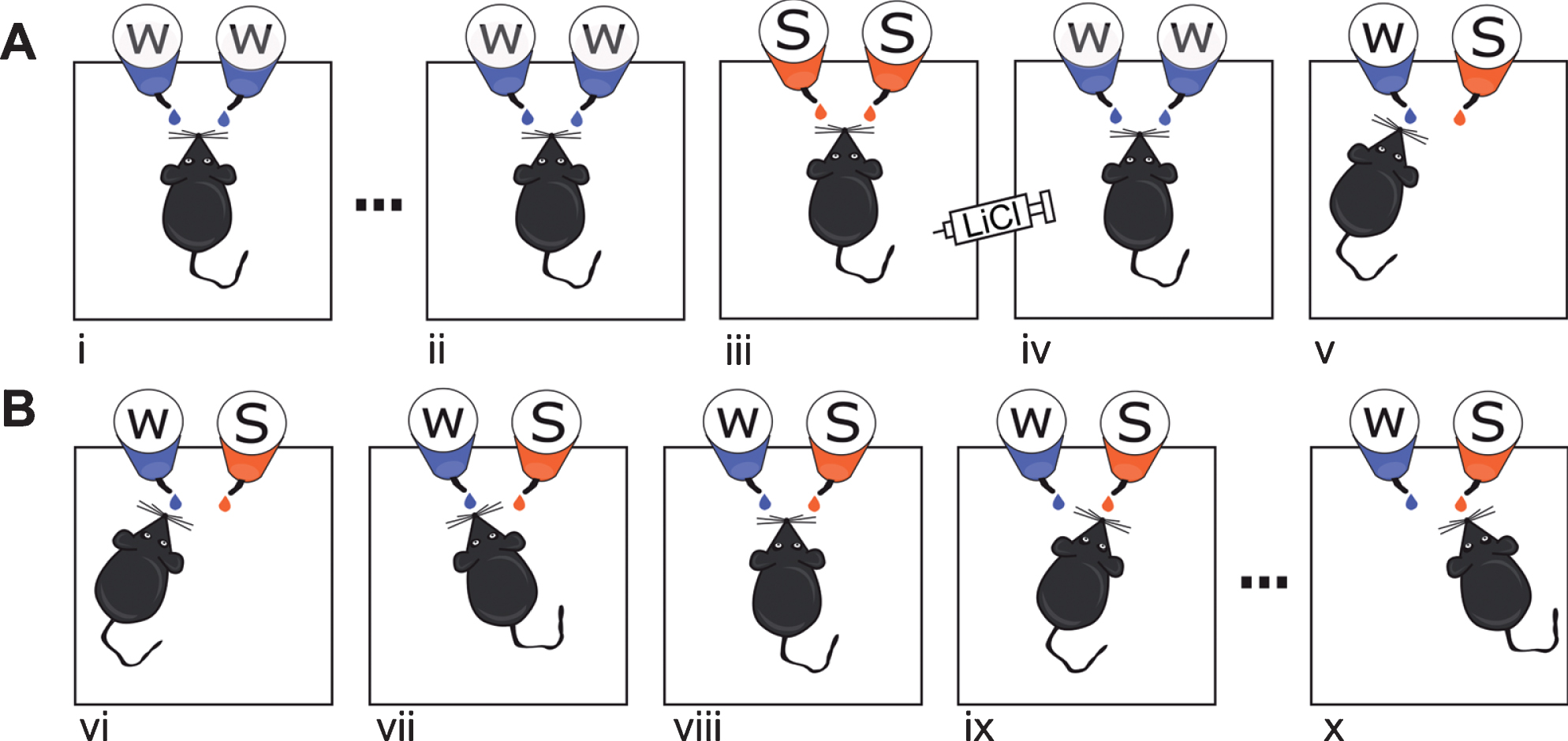
Experimental procedures. A) CTA acquisition learning. Mice were habituated to receive water via two spouts over 20 min once every 12 h (i, ii); following habituation, mice were given CTA training (iii), and after another water session (iv), the CTA was evaluated (v) using two pipettes: one containing sucrose and the other water. B) CTA extinction protocol. A strong CTA protocol (as in A[i-iii]) was followed by at most 7 extinction session in which two pipettes containing sucrose and water were presented for 20 min in 12-h intervals. In successful extinction, sucrose avoidance [vi] changes gradually to sucrose preference [x].
Open-field test for measuring mice LiCl susceptibility
Mice were habituated for 30 min to the experimental room before individually placed in a 50×50×30 open-field arena, which was wiped with 70% (v/v) ethanol before each test to minimize olfactory cues. The trial started with 1 h of habituation followed by an IP injection of 0.15M LiCl at 2% of body weight and immediately put back into the arena for an additional hour. Mice movements were video captured and processed using Ethovision XT (Noldus; Wageningen, Netherlands). The summed movement of each mouse in the 30-min post injection was normalized with its summed movement in the 30-min prior to the injection.
Hot plate test for measuring mice pain thresholds
Each mouse was individually placed on a hot plate apparatus (Ugo Basile, Italy, Hot/Cold Plate NG) covered by a transparent cylinder (25 cm height). For each mouse, the hot plate was initially set to 19°C±0.2 and gradually increased. Mice were removed once a clear paw liking movement or jumping indicating response to heat was detected, and the temperature was recorded.
Sucrose detection test
Mice were placed in individual cages and their regular bottles were replaced with two 10 ml surgical pipettes. During the first and second days both pipettes contained water for habituation. During the experiment trials, each lasting 24 h starting from 9AM of each day, mice received two different randomly placed pipettes containing water and sucrose solution. The sucrose solution placed in each trial contained decreasing concentrations of sucrose: 100 mM, 50 mM, 25 mM, 12 mM, and 6 mM solutions. The consumption from each pipette was measured daily.
Tissue collection, molecular procedures, and microscopy
Mice were deeply anesthetized with isoflurane 90 min upon the termination of the experiment and perfused with phosphate buffer (PBS) followed by 0.01 M PBS containing 4% paraformaldehyde. Brains were obtained and postfixed in 30% sucrose-paraformaldehyde for 3 consecutive days. Coronal sections were serially cut at 50μm using a freezing sliding microtome. BLA, GC, hippocampal CA3, and mPFC containing slides were used for immunohistochemistry with primary antibodies for: vesicular glutamate transporter (vGlut), vesicular GABA transporter (vGat), and Synaptophysin (Rabbit anti-vGlut1: Abcam 1:1000; Rabbit anti-vGat: Abcam 1:1000 and Rabbit anti-Synaptophysin: Abcam 1:2000).
Immunohistochemistry
Sections were first washed 3 times with 200μl PBS and then 3 additional times with PBS solution containing 0.1% Triton X-100 (PBST). The sections were then maintained in 20% goat serum (normal goat serum, Cell Signaling) for 1.5–2 h at room temperature. Goat serum was replaced by 3 washes of PBST and sections were incubated with a primary antibody for 30 min at 37°C and then maintained at 4°C for 48 h. Subsequently the primary antibody was washed 3 times with PBST and replaced by the corresponding secondary antibody (Cell Signaling Technology, Danvers, MA) diluted in PBST for 1 h protected from light at room temperature. Finally, sections were washed again with PBST and mounted on a dry slide, dehydrated and sealed with DAPI mounting solution (Invitrogen, Carlsbad, CA).
Brain slices were visualized using a confocal scanning laser microscope (Zeiss, LSM 510, Axioskop, Oberkochen, Germany). Three captions were taken from each brain region (GC, BLA, mPFC, and hippocampus) of each animal under similar conditions. Intensity calculations of the immunofluorescence staining were carried by the ImageJ software without further procedures for the GC, BLA, and mPFC containing sections. Hippocampal sections from bregma –1.7 to 2.10 were marked to select the CA3 and a threshold was set specifically for each type of primary antibody, as previously done by [12]. The microscopy acquisition was conducted in a blinded manner followed by a computerized automated analysis to reduce any bias.
Statistical analyses
The results in the graphs are expressed as means±SEM. Repeated measure two-way ANOVA was performed for comparing the differences between the groups across learning. A paired t-test was performed to assess within-group changes. Molecular differences were tested using two-way ANOVA. The value in parenthesis of the F and t statistics represent degrees of freedom. The significance level was set to 0.05 for all statistical analyses. Since we did not find any difference in consumption between females and males within the same genotype across all tests the results of males and females were pooled together.
RESULTS
CTA acquisition learning is impaired in young apoE4 mice
We first tested for differences in the ability of the two genotypes to acquire CTA. To avoid a floor effect that may obscure differences between the genotypes, a weak CTA procedure was conducted. The apoE4 and apoE3 mice consumed comparable amounts of water in the last habituation trial (t-test, t(20) = 14.6, p = 0.87) and sucrose (t-test, t(20) = 0.61, p = 0.89) during the CTA training session. Both groups significantly increased sucrose consumption in the training day compare to water consumption in the previous day (Paired t-test, apoE3: t(10) = 6.05, p < 0.001; apoE4: t(10) = 3.84, p < 0.01). These results indicate similarity in the hedonic value perception of sucrose and an expected lack of sucrose neophobia (fear of novel foods) in both groups [34]. Significant differences between the groups, however, were found in their ability to learn the taste-malaise association tested a day later. While the apoE3 mice showed a strong avoidance reaction towards sucrose, apoE4 mice demonstrated high sucrose preference (Fig. 2A; t-test, t(20) = 4.7472, p < 0.001). We further verified that this difference was not due to alterations in the perception of either the taste or the malaise. Specifically, both genotypes showed a similar preference for a range of sucrose concentrations, demonstrating a similar sensitivity to sucrose (Fig. 2B; Repeated measurements two-way ANOVA, Genotype F(1,2) = 0.14, p = 0.73, Interaction F(4,8) = 0.34, p = 0.49). Both groups also showed similar general pain sensitivity in the hot plate test (Fig. 2C; t-test, t(7) = 0.7639, p = 0.466) and similar reduced locomotion in response to gastric malaise alone tested in the open field as an indirect measure of their malaise perception (t-test, t(5) = 1.313, p = 0.228). Taken together, these results suggest an impaired capacity of apoE4 mice to acquire CTA under weak conditioning.
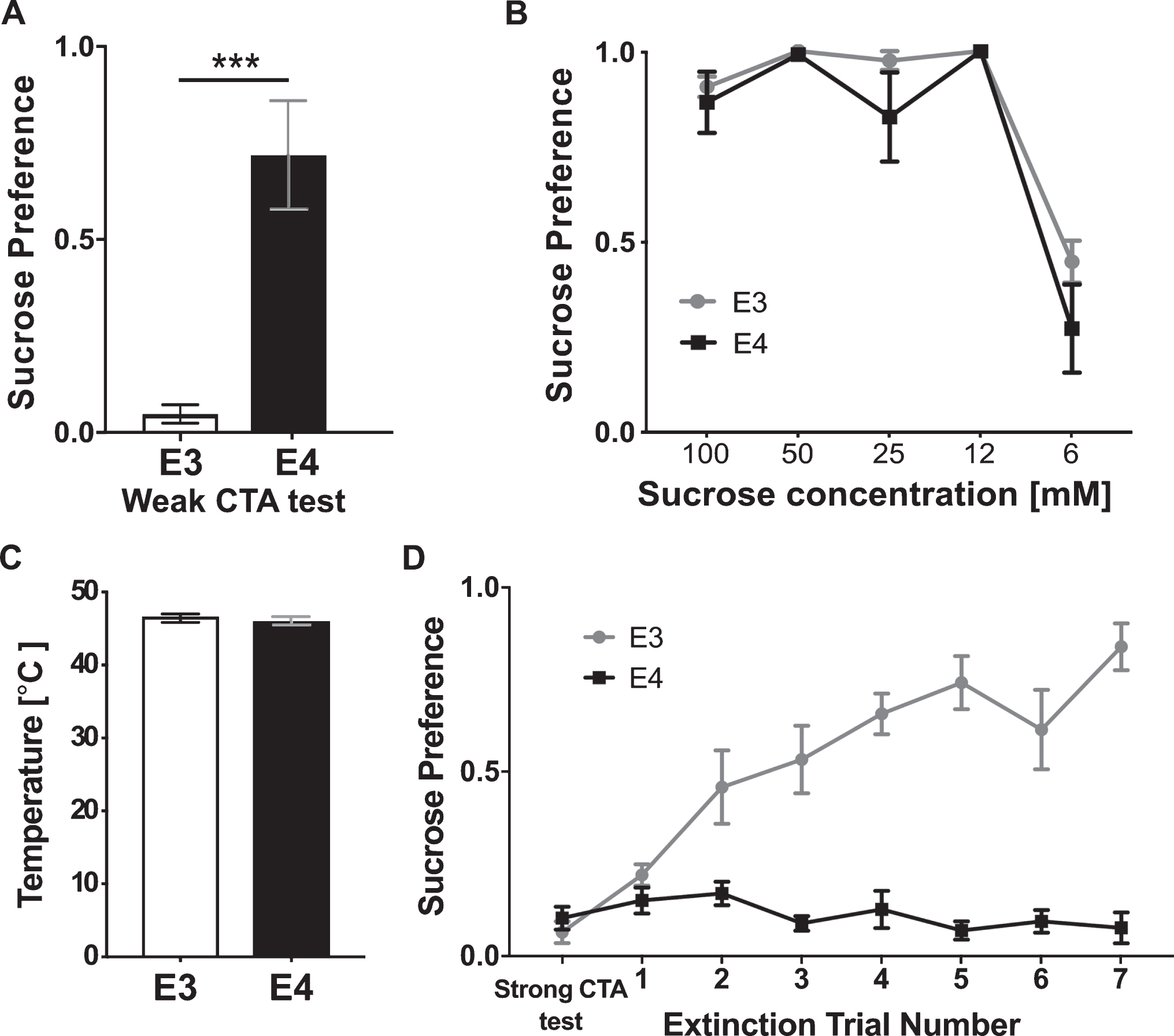
CTA acquisition and extinction are impaired in apoE4 mice. A) Weak CTA caused pronounce sucrose aversion in apoE3 mice, while apoE4 mice failed to acquire CTA and showed high sucrose preference. B) ApoE3 and apoE4 mice show similar preference to decreasing sucrose concentrations. C) ApoE3 and apoE4 mice show similar sensitivity to nociceptive pain measured in the hot plate test. D) Following strong conditioning all genotypes successfully avoided the sucrose; however, the ApoE4 group was unable to extinguish the association, while apoE3 mice did. E3, apoE3 mice; E4, apoE4 mice. The graphs represent the mean±SEM; A, B n = 11 per group; C, D n = 4-5 per group; ***p < 0.001.
Young apoE4 mice show extinction deficits of an acquired CTA memory
Next we examined whether apoE4 also impairs CTA extinction learning. A higher dose of LiCl (0.15M LiCl at 2% of bodyweight) was used to induce a strong and similar aversion of the apoE4 and apoE3 mice to sucrose (Fig. 2D, t-test, t(20) = 0.89 p = 0.3805) to allow testing of extinction rate from a similar initial condition. The results from the two-pipette tests (see Methods) conducted in 12-h intervals show that while apoE3 mice have gradually increased their preference for sucrose over 3 sessions, apoE4 mice maintained stable low sucrose consumption during the entire period of extinction training (Fig. 2D, Repeated measurements two-way ANOVA; Genotype effect F(1,160) = 221.2, p < 0.0001, day effect F(7,160) = 10.17, p < 0.0001, interaction F(7,160) = 13.23, p < 0.001). Importantly, the impaired extinction was not due to a lack of sucrose exposure since apoE4 mice have sampled the sucrose pipette during each trial. These results, therefore, suggest an apoE4-related CTA extinction learning deficit.
Impaired CTA acquisition in young apoE4 mice correlates with altered pre-synaptic plasticity markers in the GC and BLA
Following the marked learning impairments, we tested whether the structural synaptic deficits in the GC and BLA may account for the impaired CTA acquisition. Reduced basal vGat levels were found in the apoE4 compared to apoE3 mice only in the BLA (Fig. 3A). Weak CTA, however, caused an increase in vGat levels in apoE3 mice in both the GC and BLA, but significantly less in the apoE4 mice (Fig. 3A,B). The strong CTA evoked comparable vGat levels between the groups matching its induction of comparable avoidance behavior (Fig. 3A,B). In contrast, CA3 vGat levels showed no response to any of the behavioral tasks (Fig. 3C). Thus, vGat levels are finely correlated with the behavior showing reduced levels in apoE4 mice relative to the apE3 mice when weak CTA acquisition is impaired, and fully elevated levels when apoE4 mice manage to achieve strong CTA.
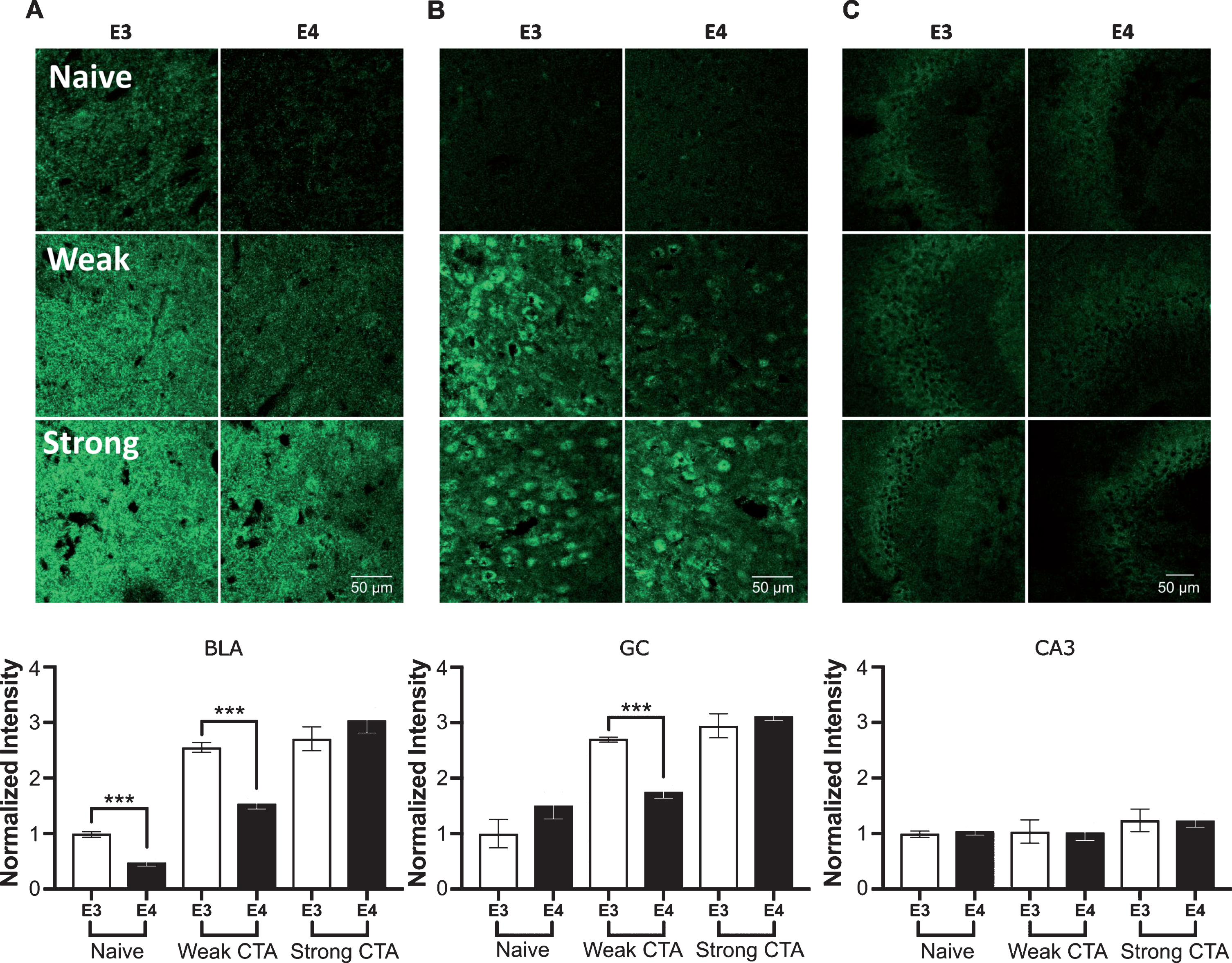
vGat structural plasticity changes in the GC, BLA, and CA3 following CTA learning. A) BLA basal vGat levels were lower in apoE4 compared to apoE3 mice, and similarly to the GC showed impaired increase with weak CTA and normal levels following strong CTA. B) ApoE3 mice increased their GC vGat levels from their initial naïve levels in a response to weak CTA, whereas apoE4 have not. Both groups showed similar higher densities following strong CTA. C) The CA3 was not affected by the tested behavioral paradigms. E3, apoE3 mice; E4, apoE4 mice. Upper panels – representative sections. Lower panels show the population quantized levels normalized to the apoE3 naïve group of each brain region. The graphs represent the mean±SEM; n = 5-6 per group; ***p < 0.001.
Glutamatergic pre-synaptic vesicular levels were detected using an antibody for vGlut. As in vGat, basal vGlut levels were significantly lower in apoE4 than apoE3 mice only in the BLA (Fig. 4A). The vGlut response to CTA learning resembled the task driven phenotype portrayed by vGat. Specifically, following weak CTA, vGlut levels were lower in the GC, BLA, and CA3 of apoE4 compared to apoE3 mice (Fig. 4). Moreover, in the GC, weak conditioned vGlut apoE4 levels remained similar to their naïve levels (Fig. 4A) (t-test, t(20) = 0.89, p > 0.1). In contrast, no significant vGlut differences were found between the genotypes following strong CTA in the BLA, CA3, and the GC.
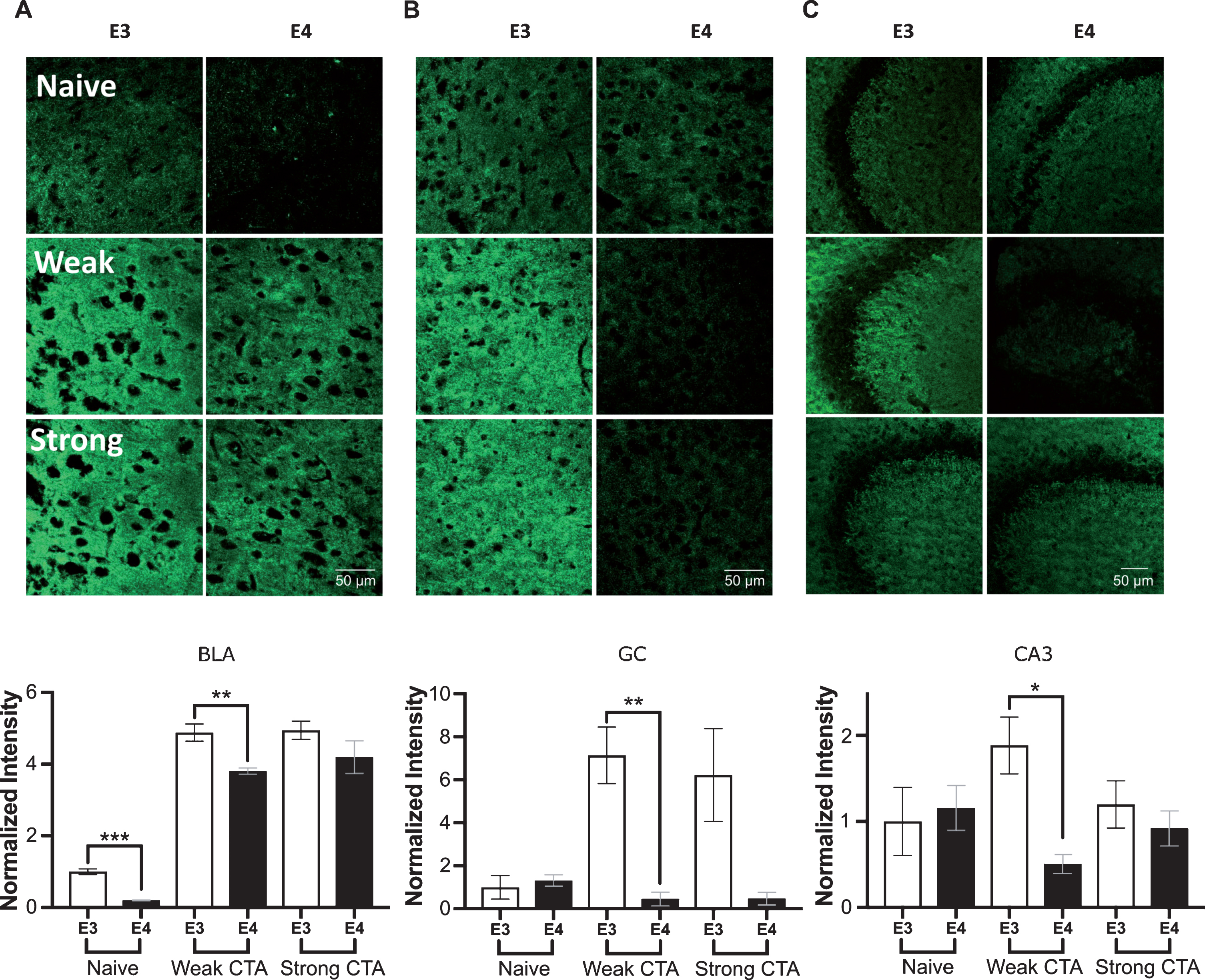
vGlut plasticity changes in the GC, BLA, and CA3 following CTA learning. A) BLA basal vGlut levels were higher in apoE3 compare to apoE4 mice. Following weak CTA apoE3 mice showed increase in vGlut, which was less pronounced in apoE4 mice. Strong CTA affected a similar increase in both genotypes. B) GC weak and strong conditioning caused a sharp increase in apoE3 vGlut level but reduced levels in apoE4 mice. C) CA3 vGlut levels increased in apoE3 group following a weak CTA, but decreased in the apoE4 group. Strong CTA drove vGlut expression in both genotypes to a similar level. E3, apoE3 mice; E4, apoE4 mice. Upper panels – representative sections. Lower panels show the population quantized levels normalized to the apoE3 naïve group of each brain region. The graphs represent the mean±SEM; n = 5-6 per group; *p < 0.05; **p < 0.01; ***p < 0.001.
The similarity between vGlut and vGat task-driven patterns may indicate a more general disturbance in task-related plasticity, which is not specific to a particular type of synapse. To examine this assertion, we examined the general marker for pre synaptic nerve terminals, synaptophysin. Basal differences in naïve animals were only observed in the GC (Fig. 5B). Like vGat and vGlut, apoE4 animals showed an impaired increase in synaptophysin levels in the BLA after weak CTA (Fig. 5A). In the GC, the behavioral paradigm effect on synaptophysin levels was particularly complicated: the initial naïve apoE4 lower levels were increased by the weak CTA reaching similar values to the apoE3 group, but again were significantly lower following strong CTA (Fig. 5B). Surprisingly, this effect of strong conditioning, repeated also in CA3 (Fig. 5C). Overall, synaptophysin levels showed less correlation with behavior compared to vGat and vGlut.
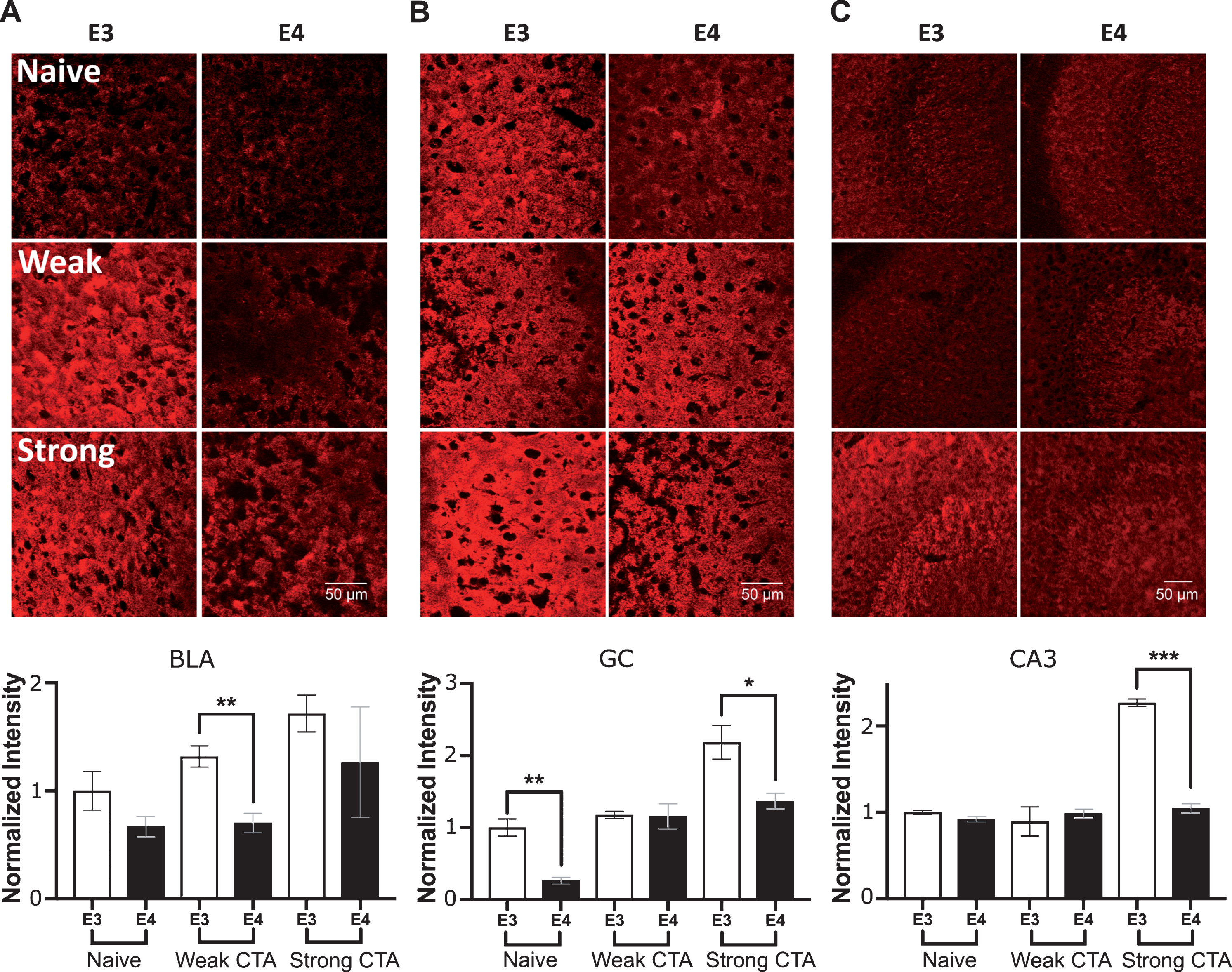
Synaptophysin plasticity changes in the GC, BLA, and CA3 following CTA learning. A) In the BLA, apoE3 animals showed an elevated level of synaptophysin after weak conditioning, but not in the apoE4 group. B) In the GC, apoE4 animals show reduction in Synaptophysin in both naïve animals and after strong conditioning. C) CA3. Synaptophysin level was only elevated after strong conditioning. E3, apoE3 mice; E4, apoE4 mice. Upper panels – representative sections. Lower panels show the population quantized levels normalized to the apoE3 naïve group of each brain region. The graphs represent the mean±SEM; n = 5-6 per group; *p < 0.05; **p < 0.01; ***p < 0.001; #0.05 < p < 0.06.
No apoE4-related vesicular deficits are observed in the GC, BLA, and CA3 after extinction
Behaviorally, the groups differ considerably in their ability to extinguish the previously acquired CTA learning (Fig. 2E). We further tested whether the same apoE4-related plasticity markers abnormalities found after weak CTA acquisition also correlate with impaired extinction. It is important to note that the biochemical analysis of the strong CTA extinction protocol must take into account the results obtained by the strong CTA itself (Figs. 3–5). Overall, most of the tested markers showed similar levels between apoE3 and apoE4 mice following strong CTA (Apart from synaptophysin in the GC and CA3, Fig. 5B, C). We have used this similarity between the groups following strong CTA as reference for the extinction results. Therefore, the results obtained from each group following 7 days of extinction were normalized to the levels obtained from the matched group following strong CTA (see Methods). Figure 6A-C represents the intensity of the different markers following extinction normalized to strong CTA intensity values of the respective group. Surprisingly, although the apoE3 and apoE4 groups showed marked behavioral differences following extinction, most of the markers level have not changed significantly from their strong CTA levels (values close to 1). Moreover, none of the markers exhibited a difference between the apoE4 and apoE3 groups across all three brain areas (Fig. 6A-C). Note that the similarity to strong CTA levels also implies that the differences in synaptophysin levels found following strong CTA in the GC and CA3 (Fig. 5B, C) remained following extinction, and could contribute to the differences in extinction behavior. Overall, these results suggest a general stagnant biochemical state over extinction trials in the GC, BLA and CA3.
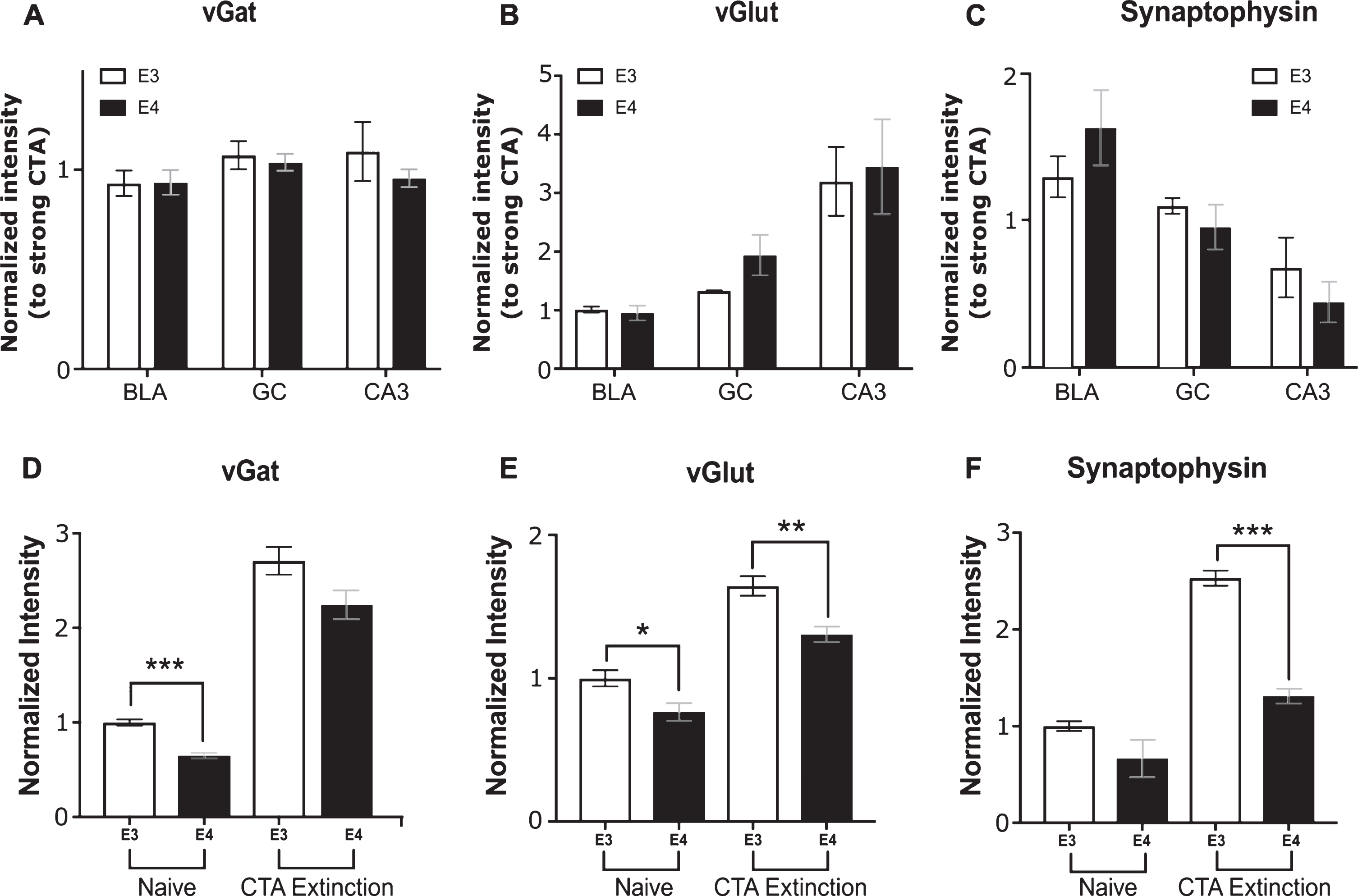
Markers levels following extinction in the GC, BLA, CA3, and mPFC. Levels of vGat (A), vGlut (B), and Synaptophysin (C) following 7 sessions of extinction. No differences were found between apoE3 (white bars) and apoE4 (black bars) when normalized to the strong CTA results of the respective genotype in the GC, BLA, and CA3. Additionally, most values remain near 1 indicating similarity with their corresponding strong CTA group levels. D-F) Markers levels in the mPFC of naïve mice and following 7 extinction sessions. In naïve mice, apoE4 mice showed reduced levels of vGat (D) and vGlut (E). Following 7 extinction days, both Synaptophysin and vGlut staining showed significant differences between the genotypes in the mPFC. E3, TR apoE3 mice; E4, TR apoE4 mice. All results are normalized to the apoE3 naïve group of each brain region. The graphs represent the mean±SEM; n = 5-6 per group; *p < 0.05; **p < 0.01; ***p < 0.001.
ApoE4 CTA extinction deficits correlate with a reduction in pre synaptic plasticity markers in the mPFC
Since no biochemical differences were detected in vGlut and vGat following extinction learning, we hypothesized that testing the mPFC, a brain region that is strongly involved in this kind of learning, may unravel additional extinction related differences between the genotypes [35, 36]. Similar to the results from the BLA, lower basal levels of vGat and vGlut were found in the mPFC of apoE4 mice compared to apoE3 mice (Fig. 6D, E). Although both groups showed elevated levels following extinction over all markers, the increase was significantly lower in the apoE4 compared to apoE3 mice in vGlut (Fig. 6E) and synaptophysin (Fig. 6F). The correlation between impaired extinction behavior and mPFC biochemical results further supports basal pre-synaptic plasticity deficits in apoE4 animals that are observed under specific conditions in different brain regions.
DISCUSSION
Young apoE3 and apoE4 mice were used to study early apoE4-related learning deficits and their pre-synaptic correlates utilizing the extra-hippocampal taste memory system of CTA. Our results show that young apoE4 mice fail to acquire a weak CTA response compared to similarly treated age-matched apoE3 mice (Fig. 2A) while strong CTA was equally learned by both genotypes (Fig. 2E). ApoE4 mice also displayed impaired extinction of a previously acquired strong CTA (Fig. 2E). Control experiments eliminated the possibility that the apoE4 mice behavioral impairments were due to differences in taste and malaise perceptions (Fig. 2B-D). Further pre-synaptic plasticity measurements in brain regions known to support CTA acquisition and extinction revealed deficits that were correlated with the observed apoE4 learning anomalies. Specifically, apoE4-related impaired CTA acquisition was correlated with reduced levels of pre-synaptic GABAergic and glutamatergic vesicles in the BLA and GC of apoE4 compared to apoE3 mice (Figs. 3A, B and 4A, B). The levels of vGlut and vGat following strong CTA were similar between the genotypes and correlated with similarity in avoidance behavior. Strong CTA related apoE4 abnormalities were observed in synaptophysin in the GC and CA3 (Fig. 5B, C) relative to those observed in the apoE3 mice. These post-strong-CTA synaptophysin anomalies remained unaltered after 7 extinction trials (Fig. 6C), and could contribute for the impaired extinction behavior. Additionally, extinction-related anomalies were also found in the mPFC— a brain region central to extinction learning— showing decreased levels of vGlut and synaptophysin (Fig. 6E, F, respectively) in apoE4 compared to apoE3 mice. The above findings are summarized in Table 1. Together, our results support the hypothesis of early apoE4-related plasticity impairments that are task-dependent and brain-region independent.
A summarizing table of the main immunohistochemical findings
A summary of the differences between the apoE3 and apoE4 genotypes in vGlut and vGat in the different experimental groups as conducted in this study. These comparisons are presented across the tested brain regions (GC, BLA, CA3, and mPFC). This model shows that when differences between apoE4 and apoE3 animals are identified, they always demonstrate lower presynaptic marker levels in the apoE4 groups.
Much of the current apoE4 (and AD) research focuses primarily on pathologies and behavior impairments related to the hippocampus. This is not surprising given the early appearance of AD in the hippocampus, and the central role of the hippocampus in memory and learning. However, if indeed apoE4 interferes with normal synaptic plasticity mechanisms as hypothesized [14–16], it should influence any brain region that requires synaptic plasticity resources. Our findings fit this task-driven perspective, showing that when an extra-hippocampal memory system employs plasticity resources, such as in the GC and BLA in weak CTA acquisition and in the mPFC through the extinction of strong CTA both the behavioral and molecular phenotypes are impaired. Interestingly, when the learning system is driven by a strong stimulus, like strong CTA, most of these differences between the genotypes are diminished. Together, these results suggest that apoE4 impairs plasticity mechanisms underlying CTA. These novel insights are the outcome of utilizing CTA paradigms which provide an easy calibration of task difficulty using a simple change in LiCl concentration affecting the levels of conditioning.
Non-hippocampal learning was only rarely studied in the scope of AD and apoE4. In humans, AD patients showed impaired acquisition and extinction of associative (non-contextual) fear conditioning [37], which is mainly subserved by the amygdala. In animal studies, apoE4 was shown to impair fear conditioning, but mainly when spatial memory [11, 12] is required, pointing again to hippocampal impairment. Our results showing impaired learning in non-contextual CTA and its extinction, memory systems that rely on extra-hippocampal brain regions, suggest that apoE4 impairments are task-dependent rather than location-specific.
Early-age assessments of behavioral and cognitive differences between the three apoE alleles showed inconsistent results [7, 38–41]. It is worth noting that most tests that were conducted to measure cognitive performance at an early age were geared towards assessing dementia-related parameters, such as working memory, language skills, reasoning, and visual perception. In contrast, our experiments were specifically designed to assess neuronal plasticity in learning over longer time periods [25, 26]. It is possible that adapting tests that rely on long term synaptic plasticity for apoE4 human subjects may reveal similar signs of early deficits as presently discussed.
Previous studies have reported a role of the GC [42, 43] and BLA [27, 44] in CTA extinction; however, our results did not show vGlut and vGat differences between the genotypes (Fig. 6A,B). This lack of differences can be explained by either reduced requirement for plasticity in these regions during CTA extinction compared to acquisition (as was previously shown [27]), or that differences exist in other factors. The former explanation suggests that other brain regions, such as the mPFC, may be more involved in extinction learning and therefore more vulnerable to the apoE4 plasticity deficits. This is backed by the specific apoE4 vGlut decrease which was detected in the mPFC following extinction. The later explanation is further supported by our data showing GC extinction-related apoE4 anomalies in synaptophysin (Figs. 5B, 6C) which may be accounted by additional vesicles other than vGat and vGlut [45].
Our results are in line with the hypothesis that apoE4 impairs learning processes through interruption with neuronal plasticity [14, 46]. Mechanistically, previous studies have shown deficits in related biochemical mechanisms, such as impaired lipid transport and metabolism [47, 48], impaired intercellular signaling [46, 49], and impaired vesicular flux [14, 51]. These cellular functions when impaired can cause a malfunction in the learning-required synaptic plasticity under load [10, 16], which might explain the results obtained in our study.
The present study introduced highly robust behavioral paradigms that can serve for the study of the mechanisms underlying primary apoE4 abnormalities. A better understanding of the early apoE4 dysfunctions may aid in the development of therapeutic targets that may contribute to the treatment and prevention of AD in apoE4 carriers. Additionally, modifications of CTA paradigms can be developed as preclinical testing of young human apoE4 carriers.
Footnotes
ACKNOWLEDGMENTS
We would like to thank the Myers Neuro-Behavioral Core Facility of Tel Aviv University for assistance with the open field and hot plate tests in this study. Special thanks to Dr. Alexander Barbul for his help with operating and obtaining images from the Zeiss microscope. This work was supported in parts by grants to DMM from the Israel Science Foundation (grant #794/17), the Joseph Sagol Foundation, from the Harold Eleanore Foonberg Foundation, and the Joseph K. and Inez Eichenbaum Foundation. In addition, the work was also supported by grants to AM from the Leir Foundation and The National Institute of Psychobiology in Israel.