
Abstract
Chronic inflammatory diseases, while seriously impairing patients' quality of life, are a heavy financial cost to the National Health Service (NHS) and to society. The availability of biological drugs – among which infliximab (Remicade®) – greatly improved treatment efficacy. On the other hand, these drugs are an expensive resource. Infliximab patent protection is going to expire, and a biosimilar has been recently approved.
A budget impact (BI) analysis was conducted to evaluate the favourable consequences – for the Italian NHS – of the biosimilar availability in terms of cost containment (savings), thanks to its lower price compared to the originator's. The analysis model expects that some patients in treatment with the originator will switch (according to a prudent assumption of the market uptake rate) to the biosimilar and that many naïve patients will directly start treatment with the biosimilar (according to a bolder uptake rate assumed). Separately considering all the different diseases for which infliximab is indicated, the number of patients who might potentially use the biosimilar is estimated – based on disease prevalence and incidence rates, the overall proportion of treated patients and the infliximab market share. The time horizon extends to five years (starting from 2015). The biosimilar price discount is 25%.
The results from the analysis show (in the base case) that the availability of the biosimilar would provide overall annual savings over €16 million to the NHS in 2019, while the cumulated savings in the five years period would be no less than €47 million. The sensitivity analysis highlights that such favourable results would be even more substantial, to the extent that switching from originator to biosimilar could be safely recommended.
Introduzione e obiettivi
Le malattie infiammatorie croniche, a eziopatogenesi autoimmune, si caratterizzano per l'importante disabilità che arrecano ai pazienti, determinando gravi conseguenze sulla qualità di vita degli stessi. Va inoltre considerato che, essendo per definizione patologie croniche, rappresentano un considerevole onere per i Servizi Sanitari Nazionali (SSN) a causa degli elevati costi di trattamento, che assorbono una parte importante delle risorse disponibili per la tutela della salute di un Paese (1).
Rientrano in questo gruppo di malattie l'artrite reumatoide (AR), la spondilite anchilosante (SA), la malattia di Crohn (MC), la colite ulcerosa (CU), l'artropatia psoriasica (AP) e la psoriasi (Ps). Il grado di disabilità di tali patologie, elevato sin dalle fasi iniziali, incide pesantemente sulla qualità della vita non solo del paziente, ma anche dei suoi familiari e più in generale delle persone che se ne occupano (2). Si tratta pertanto di malattie a forte impatto sociale; la parte prevalente dei costi di queste malattie è costituita dalle assenze dal posto di lavoro (molti pazienti abbandonano precocemente l'attività lavorativa) e dalle cure informali fornite su base volontaria dai caregiver, cioè l'assistenza fornita al paziente non contemplata dal SSN o da operatori privati (3). Si stima che i costi diretti sanitari rappresentino meno della metà del totale, come per la quasi totalità delle malattie cronico-degenerative (4–6).
L'incidenza e la prevalenza di queste malattie non sono particolarmente elevate, con l'eccezione della Ps, ma nel loro insieme rappresentano nel panorama italiano una frazione non secondaria della popolazione. Prevalenze più elevate si riscontrano, per alcune di esse, nei Paesi del Nord Europa (7). Applicando dati di prevalenza alle popolazioni di Regno Unito, Germania, Italia, Paesi Bassi e Belgio, si può stimare che quasi sette milioni di pazienti siano affetti da tali patologie.
I trattamenti farmacologici a oggi non determinano la guarigione, ma possono permettere lo stato di remissione o una bassa attività di malattia. L'avvento dei farmaci biologici sul finire degli anni novanta, e in particolare di infliximab (1999), il primo anticorpo monoclonale, inibitore del tumor necrosis factor-α (anti TNF), ha cambiato la storia del trattamento di queste patologie. Tali farmaci si sono dimostrati estremamente efficaci nel breve e nel lungo periodo. Caratterizzati da una provata efficacia e da un buon profilo di sicurezza i farmaci biologici rappresentano una delle voci di costo più rilevanti per il SSN. Secondo le attuali linee guida delle diverse società scientifiche italiane e internazionali i farmaci biologici oggi trovano indicazione solo dopo il fallimento dei farmaci di fondo tradizionali (es. metotrexato per l'AR, mesalazina e/o azatioprina per le malattie infiammatorie intestinali, ciclosporina, metotrexato o PUVA per la Ps) o in quei pazienti per i quali vi siano controindicazioni all'uso dei farmaci di fondo. È tuttavia noto che, proprio per l'elevato costo, diversi pazienti eleggibili ai biologici non hanno accesso al trattamento con questi farmaci nonostante alcuni studi farmacoeconomici abbiano mostrato, anche se non in tutte le patologie considerate, rapporti di costo-efficacia degli anti TNF entro o al limite degli intervalli di accettabilità nei confronti dei farmaci di fondo tradizionali. Questo aspetto verrà ripreso nella
L'approssimarsi della scadenza della protezione brevettuale di infliximab, prevista nel mese di febbraio 2015, ha indotto alcune aziende farmaceutiche a investire risorse nella produzione del suo corrispettivo farmaco “biosimilare”. I farmaci biologici sono costituiti da molecole complesse, il cui processo produttivo può determinare variazioni nelle loro caratteristiche molecolari e potenziali cambiamenti delle proprietà terapeutiche (8). L'European Medicines Agency (EMA) ha provveduto a emanare linee guida (“Guideline on Similar Biological Medicinal Products”) (9–11) nelle quali viene specificato l'iter a cui deve essere sottoposto un farmaco biosimilare per ottenere l'autorizzazione all'immissione in commercio: l'esercizio di comparabilità. Questo esercizio è basato su un solido confronto diretto tra il biosimilare e il medicinale di riferimento in termini di qualità, sicurezza ed efficacia clinica e non clinica. La comparabilità della qualità è determinata rispetto alla struttura molecolare oltre che rispetto alla funzionalità, e deve essere dimostrata con un'esauriente caratterizzazione analitica e con studi e test biologici sui legami dei recettori coinvolti, da eseguirsi in modo strettamente comparativo sul biosimilare e sul medicinale di riferimento. La comparabilità clinica e non clinica garantisce che eventuali differenze osservate in termini di qualità non abbiano alcun impatto sulla sicurezza ed efficacia del medicinale biosimilare rispetto al medicinale di riferimento. Nel giugno 2013 EMA ha approvato il primo biosimilare di infliximab (CT-P13) per le stesse indicazioni del medicinale di riferimento (AR, SA, MC, CU, AP e Ps). L'esercizio di comparabilità condotto per infliximab biosimilare ha previsto oltre 50 test e studi in vitro e in vivo di confronto delle caratteristiche fisico-chimiche e biologiche con infliximab di riferimento (12). La struttura primaria dei due anticorpi monoclonali è risultata identica, le strutture di grado maggiore erano indistinguibili, simile potenza e analoghe affinità recettoriali. La farmacocinetica e l'equivalenza clinica di infliximab biosimilare e del suo riferimento sono state dimostrate invece da due studi randomizzati, controllati, in doppio cieco, su pazienti con SA e AR. Il primo studio (PLANETAS) (13) è stato condotto in pazienti con SA. PLANETAS è uno studio multicentrico di fase I, randomizzato, in doppio cieco, a gruppi paralleli disegnato per confrontare farmacocinetica, efficacia e sicurezza d'uso di infliximab biosimilare al dosaggio di 5 mg/kg con quelle di infliximab di riferimento, sempre al dosaggio di 5 mg/kg, in 250 pazienti affetti da SA. Il secondo studio (PLANETRA) (14) è uno studio di fase III, randomizzato, in doppio cieco, multicentrico, a gruppi paralleli, disegnato per confrontare efficacia e sicurezza d'uso di infliximab biosimilare al dosaggio di 3 mg/kg con quelle di infliximab di riferimento, sempre al dosaggio di 3 mg/kg, in 606 pazienti con AR attiva e insufficiente risposta al trattamento con metotrexato. Il profilo farmacocinetico dei due prodotti è risultato equivalente in entrambi i trials, e nei due studi si evidenzia inoltre l'equivalente profilo di efficacia, tollerabilità e immunogenicità a 30 settimane del biosimilare rispetto a quello di infliximab di riferimento. Questi risultati sono stati confermati dai successivi dati di follow-up a 54 settimane (15, 16). Comparabile efficacia, tollerabilità e immunogenicità è stata dimostrata anche nella fase di estensione in aperto, a 48 settimane, degli studi clinici PLANETRA e PLANETAS, tra i gruppi di pazienti che hanno continuato il trattamento con infliximab biosimilare (gruppo mantenimento) e quelli che sono passati da infliximab di riferimento a infliximab biosimilare (gruppo switch) (17, 18).
EMA nel 2009 ha confermato che, sulla base delle evidenze complessive fornite dall'esercizio di comparabilità (i test fisicochimici e le analisi biologiche insieme con i risultati degli studi farmacocinetici e clinici di efficacia e sicurezza nei pazienti con AR e SA), sono state dimostrate la biosimilarità e l'equivalenza terapeutica tra i due prodotti. Sulla scorta di questi dati EMA ha approvato l'estensione delle indicazioni di infliximab biosimilare a quelle del prodotto di riferimento e cioè, oltre a AR e SA, anche a MC, CU, AP e Ps.
I farmaci biosimilari non offrono, per definizione, benefici clinici rispetto ai farmaci originatori. Il beneficio atteso dalla loro immissione in commercio è rappresentato dal loro minore prezzo, e quindi dal conseguente effetto competitivo che si può generare creando nel tempo risparmi potenzialmente consistenti nella spesa farmaceutica pubblica. È merito dei farmaci biosimilari finora introdotti sul mercato, quali gli ESA (erythropoiesis stimulating agents) e i G-CSF (granulocyte-colony stimulating factors) l'essere stati di stimolo alla competizione nel mercato dei prodotti farmaceutici ad alto costo.
Due studi di autori italiani si sono occupati dello sviluppo dei biosimilari in Italia. Il primo studio (19), pionieristico in quanto pubblicato nel 2010, ha ipotizzato per questi farmaci, pur con differenze tra le varie classi terapeutiche, un tasso di penetrazione medio in Italia del 10% massimo al primo anno di lancio e del 40% al quarto anno dalla scadenza del brevetto, e un differenziale di prezzo compreso tra il 20% e il 40% a seconda della tipologia dei prodotti. I risparmi annui così ottenibili per il SSN sono stati stimati pari a 200 milioni di euro nel 2015 e a 500 milioni nel 2020. Il secondo studio (20) ha analizzato i dati consuntivi del mercato degli ESA e dei G-CSF in cinque Paesi europei negli anni dal 2008 al 2011. Considerando il 2009 come anno di lancio, in quanto nel 2008 in Italia e in molti dei Paesi considerati non erano state registrate vendite di biosimilari, la quota di mercato mediamente raggiunta dagli ESA nei cinque Paesi considerati (Italia, Francia, Germania, Regno Unito e Spagna) è stata del 3,1% nel 2009, del 5,7% nel 2010 e dell'8,5% nel 2011; per i G-CSF è stata del 3,2% nel 2009, del 7,4% nel 2010 e del 12,7% nel 2011. In Italia nel 2011 si è arrivati al 6,6% per gli ESA e al 16,1% per i G-CSF. In questo secondo studio non sono riportate stime dei risparmi originati da queste quote di mercato.
Ancora uno studio italiano (21), confluito poi in (22), propone una stima degli effetti competitivi di prezzo dei biosimilari su epoetina, somatropina e filgrastim in Italia.
Pur in assenza di studi sistematici sui risparmi conseguiti dal SSN grazie ai biosimilari, studi di mercato indicano che essi sono in crescita, a livello nazionale e in alcune regioni italiane in particolare (23, 24). È quindi realistico prevedere che i biosimilari forniranno un importante contributo al controllo della spesa farmaceutica pubblica, grazie anche all'apporto di infliximab biosimilare, la cui immissione nel mercato italiano è prevista nel 2015.
Da ciò consegue l'obiettivo del presente articolo, che è quello di valutare quale contenimento della spesa stessa potrebbe derivare dai potenziali risparmi – Drug Budget Impact (DBI) – generati in Italia nei prossimi cinque anni dall'utilizzo del farmaco infliximab biosimilare (CT-P13, di seguito “biosimilare”). Da notare che la sovrapponibilità terapeutica rende non rilevanti le valutazioni su eventi evitati ed effetti collaterali.
Materiali e Metodi
Per valutare il DBI del biosimilare è stato messo a punto un modello, dove si prevede che tale farmaco – meno costoso del biologico originatore (Remicade®) – possa in parte sostituirsi a quest'ultimo. Per effetto di questo processo si genererebbero dei risparmi per il SSN, da quantificare confrontando la spesa a carico del medesimo nelle ipotesi, rispettivamente, di assenza e di presenza del biosimilare sul mercato. In questa seconda ipotesi, tale spesa avrà due addendi: il costo dei pazienti trattati col biosimilare e quello dei rimanenti pazienti trattati con l'originatore.
La configurazione del modello (basato su diverse ipotesi semplificatrici che saranno riprese nella Discussione) è qui delineata in quattro punti:
l'orizzonte temporale dell'analisi si estende su cinque anni (a partire dal 2015);
le indicazioni terapeutiche dei farmaci prese in considerazione sono quelle già menzionate nell
la diffusione del biosimilare è analizzata distinguendo due tipi di popolazione interessata a infliximab: quella dei pazienti già in corso di trattamento con l'originatore, per i quali l'eventuale adozione del biosimilare non potrebbe effettuarsi che col passaggio da quello a quest'ultimo (switch population); e quella dei pazienti che si affacciano per la prima volta al trattamento (naïve population), nel qual caso il biosimilare potrà essere adottato in partenza al posto dell'originatore. Il merito di tale distinzione è di consentire un'analisi più realistica, permettendo di ipotizzare la conquista di quote di mercato (uptake) [riferite, in questo contesto, al numero di pazienti] da parte del biosimilare che sono diverse a seconda della popolazione considerata: per il tasso di uptake nella switch population sarà infatti assunto un valore più prudente rispetto a quello nella popolazione naïve, in sintonia con l'atteggiamento tenuto in generale dai clinici in fatto di sostituzione tra farmaci biologici.
Ciò premesso, per il dato della popolazione italiana residente nel 2015 è qui adottata la previsione ISTAT (25). La switch population è dimensionata nel primo anno in funzione del tasso di prevalenza della malattia (nella Tab. I sono specificate le fonti dei dati), della percentuale di pazienti trattati (26) e – di questi – della percentuale dei trattati con l'originatore (27); tale popolazione viene mantenuta costante negli anni successivi. Applicando a questa popolazione il tasso di uptake ipotizzato per quell'anno si ottiene il numero di pazienti passati al biosimilare nell'anno stesso (e, per differenza, di quelli mantenuti dall'originatore).
Dati epidemiologici

Diversamente, la naïve population varia (in aumento) di anno in anno, anzitutto perché il tasso d'incidenza viene applicato alla popolazione italiana ipotizzata in crescita dello 0,28% annuo (25). In secondo luogo perché, man mano, ai pazienti incidenti nell'anno si cumulano quelli degli anni precedenti (nel frattempo diventati prevalenti). In ciascun anno, applicando ai pazienti incidenti il tasso di uptake, viene calcolato il numero dei nuovi pazienti trattati col biosimilare in quell'anno; sommando a questi quelli analogamente calcolati negli anni precedenti si ottiene il numero di pazienti trattati fin dall'inizio col biosimilare. A complemento del totale si ottengono i pazienti trattati con l'originatore.
Allo sviluppo della naïve population (mentre si è visto che la switch population rimane costante) è peraltro stato dato un vincolo: che la crescita della popolazione complessiva (combined population, somma della popolazione switch e di quella naïve) risulti del 3% all'anno, cioè allineata col trend di mercato a quantità dell'originatore tra il 2011 e il 2014 (28).
La Tabella I riporta, per ciascuna malattia, i dati epidemiologici (prevalenza e incidenza) utilizzati per dimensionare i due tipi di popolazione (rispettivamente: switch e naïve). Quando non erano disponibili dati direttamente riferiti all'Italia, sono stati utilizzati come proxy quelli di Paesi confrontabili.
Indistintamente per tutte le indicazioni terapeutiche, i tassi di uptake ipotizzati nel modello (per il caso base) sono riportati nella Tabella II. Specifichiamo ancora che questi tassi sono riferiti ai pazienti, non alle vendite – anche se è comunque intuitivo un certo parallelismo tra le due variabili;
Tassi di uptake del biosimilare vs l'originatore

il costo medio di trattamento per paziente/anno è basato sul prezzo di listino dell'originatore (prevedendo un certo discount per il biosimilare) e sul numero di vials necessari per il carico e il mantenimento.
Specificamente, il costo di 1 vial di Remicade® da 100 mg è posto a €515 (33), con un discount per il biosimilare ipotizzato del 25%. Nella Tabella III sono riportate le quantità di infliximab necessarie per il trattamento annuo di un paziente.
Risorse farmacologiche di trattamento

Un'analisi di sensibilità è stata effettuata tenendo presente il caso di un orientamento pubblico volto a facilitare il passaggio dall'originatore al biosimilare: il governo norvegese ha promosso uno studio di approfondimento ad hoc (lo studio NOR-SWITCH) (35, 36), che si prevede sarà completato nella seconda metà del 2016 (37). In linea con tale orientamento, il parametro qui fatto variare (in aumento) è il tasso di uptake nella switch population, per esplorare le potenzialità del suddetto orientamento in termini di economia di spesa (a parità di efficacia e sicurezza sotto il profilo clinico).
Risultati
L'analisi di DBI del biosimilare è stata fatta a livello delle singole indicazioni terapeutiche. Viene qui di seguito presentato il DBI nel caso base; a seguire, un'analisi di sensibilità.
La Tabella IV è relativa all'AR, di seguito sono presentate le tabelle per le cinque rimanenti indicazioni (Tabb. V-IX). La successiva Tabella X costituisce un'aggregazione di tutte le sei tabelle analitiche.
Drug Budget Impact di infliximab biosimilare nell'artrite reumatoide

Drug Budget Impact di infliximab biosimilare nella spondilite anchilosante

Drug Budget Impact di infliximab biosimilare nella malattia di Crohn

Drug Budget Impact di infliximab biosimilare nella colite ulcerosa

Drug Budget Impact di infliximab biosimilare nell'artrite psoriasica

Drug Budget Impact di infliximab biosimilare nella psoriasi

In tutte le tabelle, per consentire una rappresentazione leggibile nell'ambito di una pagina di testo, sono riportati solo l'anno iniziale e finale del quinquennio considerato nel modello.
Sulla base dei dati epidemiologici della Tabella I, la prevalenza dell'AR in Italia all'inizio del 2015 è stimabile in 203.402 pazienti. Di questi, 1180 sarebbero trattati con l'originatore (26, 27).
Nello stesso anno, i pazienti incidenti sarebbero 6164, di cui 36 trattati con l'originatore; partendo da questi, per effetto dei successivi subentri si arriverà a fine quinquennio a una naïve population di 188 pazienti.
Assumendo che nel 2015 il biosimilare sia immesso sul mercato – e che si realizzino i tassi di uptake ipotizzati per la switch population (Tab. II) – nel 2019 passeranno al biosimilare 295 pazienti (contro i 59 nel 2015) prima trattati con l'originatore. Mentre nel 2019, fra i trattati ex novo, 150 lo saranno col biosimilare (e solo 38 con l'originatore). In tutto (combined population), si prevede che a fine quinquennio 445 pazienti saranno trattati col biosimilare e 923 con l'originatore.
In termini di DBI, al SSN il costo complessivo per trattare questi 1.368 pazienti risulterebbe, se il biosimilare non fosse disponibile, pari a €13.874.868. Costo che si ridurrebbe a €12.737.773 grazie al minor prezzo del biosimilare, qualora questo si sostituisse/affiancasse parzialmente all'originatore nel trattamento dell'AR. In questo caso, il risparmio annuo realizzato dal SSN nel 2019 sarebbe pari a €1.137.094 e quello complessivo nel quinquennio ammonterebbe a €3.228.811. In altre parole, a ogni paziente trattato col biosimilare corrisponderebbe un risparmio annuo di €2.535.
In Italia, per tutte le patologie considerate in questo studio (Tab. X) la prevalenza risulta complessivamente pari a 2.268.242 pazienti (di cui 10.324 trattati con l'originatore). Le patologie a maggiore prevalenza sono, insieme all'AR (203.402 pazienti, Tab. IV), I'AP (117.110 pazienti, Tab. VIII,) e la Ps (1.787.473 pazienti – vale a dire, quasi i quattro quinti del totale – Tab. IX).
Drug Budget Impact di infliximab biosimilare nell'insieme delle patologie considerate
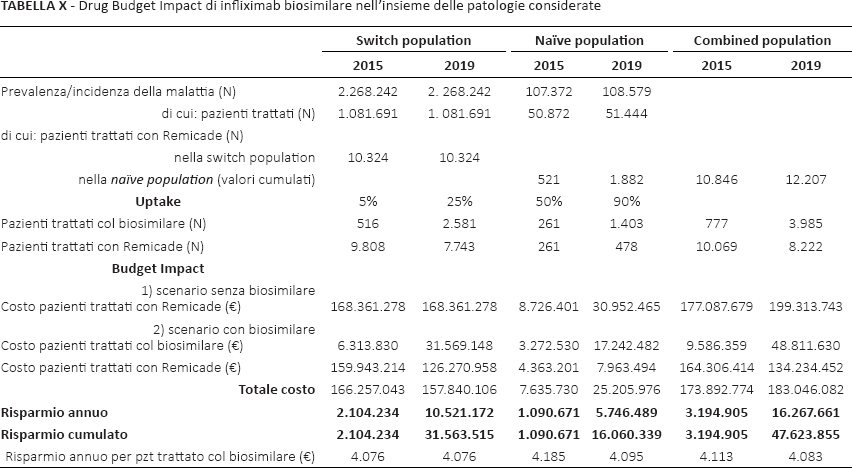
L'incidenza, nel 2015, è di 107.372 pazienti, di cui 521 trattati con l'originatore.
Il costo di trattamento complessivo di tali patologie con infliximab (con riferimento alla combined population, nel 2015, nell'ipotesi di impiegare esclusivamente l'originatore) ammonta a €177.087.679. A questo riguardo, le patologie a maggiore costo sono la CU (€80.687.149, Tab. VII), l'AP (€28.478.649, Tab. VIII) e la Ps (€31.013.724, Tab. IX). Per inciso, lo scarso parallelismo tra costi e prevalenze in tali patologie si può spiegare con la non linearità fra tassi di prevalenza e quote di pazienti effettivamente trattati con infliximab.
Il DBI del biosimilare porterebbe a €16.267.661 il risparmio annuo complessivo nell'ultimo anno del quinquennio. Sempre nell'insieme delle patologie, il risparmio annuo per ogni paziente trattato col biosimilare non sarebbe mai inferiore ai 4.000 Euro.
Se si considera, in generale, che ai costi per farmaci sostenuti dal SSN corrispondono dei fatturati incassati dalle aziende fornitrici, risulta che nel 2019 l'uptake (in termini di fatturato) del biosimilare sarebbe complessivamente di €48.811.630 su €183.046.082, cioè del 26,7% (contro il 5,5% a inizio quinquennio).
Nell'analisi di sensibilità, in seguito a quanto premesso nei
Ipotizzando di raddoppiare l'uptake del biosimilare nei pazienti già in corso di trattamento con infliximab originatore (Tab. XI), nel 2019 tutti i pazienti (combined population) di tutte le patologie trattati col biosimilare sarebbero 6566 (contro i 3985 del caso base). Nello stesso anno, il risparmio annuo complessivo sarebbe del 65% maggiore, passando da €16.267.661 (nel caso base) a €26.778.833.
Drug Budget Impact di infliximab biosimilare nell'insieme delle patologie considerate: analisi di sensibilità con tassi di uptake nella switch population raddoppiati rispetto al caso base

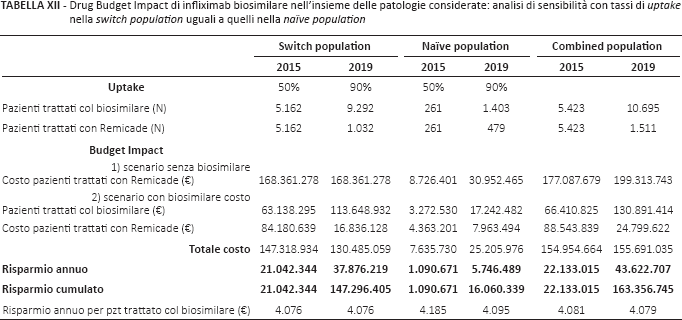
Con l'ipotesi-limite (Tab. XII) di contare sulla parità dei tassi di uptake nelle due popolazioni (di prevalenza e incidenza rispettivamente), nel 2019 il risparmio annuo quasi triplicherebbe rispetto a quello del caso base: €43.622.707 contro €16.267.661.
Drug Budget Impact di infliximab biosimilare nell'insieme delle patologie considerate: analisi di sensibilità con tassi di uptake nella switch population uguali a quelli nella naïve population
Discussione
È stata condotta un'analisi di DBI sulle conseguenze economiche per il SSN dell'autorizzazione regolatoria di un biosimilare di infliximab, ipotizzandone l'espansione sul mercato farmaceutico italiano secondo due modalità simultanee: 1) che verso il biosimilare possa esserci lo switch di una quota di pazienti già in corso di trattamento con l'originatore; 2) che il biosimilare abbia successo di prescrizioni come prima opzione in pazienti naïve.
Queste ipotesi di uptake, e la loro quantificazione nei tassi annui che si trovano alla base dell'analisi, sono – insieme al discount nel prezzo del biosimilare – in concordanza con l'ampio studio in materia di biosimilari già citato nell'
I limiti del presente lavoro sono evidenti. Il modello utilizzato per l'analisi è molto semplificato, essendo focalizzato su due sole variabili: il biosimilare e l'originatore – del quale non vengono neppure elaborati esplicitamente gli effetti di possibili strategie di contrattacco, considerati implicitamente compresi nei tassi di uptake adottati.
Altre semplificazioni da richiamare riguardano:
il tasso di uptake, che è ipotizzato identico in tutte le indicazioni: nelle due (AR e SA) per le quali sono stati fatti gli studi clinici come nelle altre;
la quota di mercato dell'originatore sul totale dei pazienti, che è assunta costante nel tempo senza considerare eventuali variazioni di mix tra l'originatore e altre molecole per la stessa indicazione;
la mancanza, nei livelli ipotizzati per i tassi di uptake, di un più solido rationale al di là della expert opinion;
il non avere incluso l'ipotesi di un effetto competitivo di prezzo sull'originatore, che avrebbe potuto portare a ulteriori risparmi nello scenario col biosimilare.
In particolare, poi, il modello in discussione non prevede che l'introduzione di infliximab biosimilare possa comportare il passaggio a infliximab di alcuni pazienti in trattamento coi DMARD (disease-modifying anti-rheumatic drug), a causa della sostanziosa diminuzione di prezzo del biosimilare. Come anticipato nell'Introduzione, alcuni studi farmacoeconomici riferiti a Paesi europei hanno confrontato i costi e l'efficacia dei due trattamenti concludendo che, nonostante il maggior costo di infliximab, il rapporto incrementale di costo efficacia (ICER) di infliximab versus DMARD si colloca entro, o poco al di sopra, il limite dell'intervallo di accettabilità proposto dal National Institute for Health and Care Excellence (NICE) di 20.000-30.000 Sterline (39). Questo però non vale per tutti gli studi analizzati. Vale, ad esempio, nel caso dell'AR, dove uno studio del 2003 riferito al Regno Unito (40) ha trovato un ICER (costo/QALY guadagnato) per infliximab vs metotrexato di £21.600 a un anno di trattamento e di £29.900 a due anni. Un ICER oltre la soglia (£36.200) è stato invece trovato da uno studio del 2011, sempre riferito al Regno Unito (41). In Italia, da uno studio del 2003 (42) si desume che infliximab sia cost-effective a partire da un valore soglia di €24.000. Nel caso della SA, uno studio (43) ha mostrato un ICER (costo/QALY guadagnato) al limite dell'intervallo di accettabilità quando infliximab viene confrontato con placebo, ma in un secondo studio (44) l'ICER di infliximab vs il trattamento convenzionale era al di sopra di tale intervallo. Nel caso della Ps, infliximab è stato confrontato anche con altri trattamenti, in particolare con etanercept. Infliximab è risultato cost-effective (ICER £26.095) rispetto a etanercept in uno studio sul trattamento della Ps severa nel Regno Unito (45), ma non in uno studio italiano (46). Nel caso dell'AP infliximab è risultato cost-effective rispetto ai DMARD (ICER £16.942-23.022) in uno studio del 2011 (47), e anche rispetto a etanercept nei casi moderati o severi (ICER £26.000) ma non nei casi lievi (ICER £65.000). Negativo invece il risultato, sempre rispetto a etanercept, in uno studio del 2006 (48) e del 2007 (49). Infine, nella CU infliximab è risultato cost-effective rispetto a ciclosporina in tre studi (50–52).
Se si considera che il biosimilare avrà un prezzo inferiore a quello dell'originatore (nel nostro studio: del 25%), sarebbe ragionevole ipotizzare che eventuali studi farmacoeconomici di confronto, quantomeno con i DMARD, potrebbero produrre risultati ancora più favorevoli, con ICER inferiori a quelli trovati nei confronti sopra riportati. Non sarebbe dunque irrealistico ipotizzare, quantomeno nel medio-lungo periodo, un possibile aumento della percentuale di pazienti trattati con infliximab biosimilare a causa di una risposta non ottimale ai DMARD convenzionali.
Comunque, anche il non avere preso in considerazione questa ipotesi (alla luce di quanto oggi previsto nelle linee guida (10)), rende conservativi i nostri risultati. Nel complesso, cioè per la maggior parte delle ipotesi, non si può dire che l'impostazione “minimalista” insita nel modello utilizzato abbia giocato a favore del biosimilare nella valutazione del suo DBI. Questo studio potrebbe anche essere un contributo metodologico all'analisi di analoghi casi di biosimilari, legati alla prossima scadenza brevettuale di altri farmaci biologici (adalimumab, etanercept, rituximab …).
Si può concludere che lo studio ha messo in luce gli apprezzabili risultati positivi che ci si potrebbero attendere in termini di risparmio nella spesa farmaceutica pubblica dall'immissione di infliximab biosimilare sul mercato. L'analisi di sensibilità indica che tali risultati sarebbero ben maggiori nella misura in cui nella prassi clinica si realizzerà lo switch dall'originatore al biosimilare, anche se questo atteggiamento deve essere attentamente valutato da un punto di vista clinico. Oltre a ciò non sarebbe da sottovalutare una possibile maggiore aderenza alle linee guida, iniziando più precocemente il trattamento biologico dei pazienti che presentano una risposta non ottimale ai DMARD convenzionali, reso possibile dal minore costo del biosimilare rispetto all'originatore.
Footnotes
Acknowledgement
The reviewer comments contributed to improve the final draft of the article.
Financial support: This research was made possible by an unrestricted educational grant from Mundipharma Pharmaceuticals Srl.
Conflict of interest: The authors declare that they have no conflict of interest related to the article.