
Abstract
3p interstitial deletions have emerged in recent years as a new cause of neurodevelopmental delay and intellectual disability. Since the first report of this condition in 1979, 16 cases have been described in the literature, delineating it as a presumptive syndrome. Here, we add a novel case presenting severely delayed neurodevelopment and psychomotor development; facial dysmorphism (square facies, broad forehead, short palpebral fissures, epicanthic folds, broad nasal bridge, and low-set malformed ears); cerebral, cardiac, and genital malformations; hand and feet anomalies; sacral sinus; and hearing impairment. Genetic investigations revealed a del(3)(p12.3p14.1) of 12.5 Mb, including 31 ORFs, among which ROBO2, PDZRN3, MITF, and FOXP1 are known to act in neurodevelopment. The clinical features of our patient are compared with those previously reported in the literature, thus providing further support for the delineation of the 3p interstitial deletion syndrome.
Among the huge number of genetic alterations associated with neurodevelopmental conditions, those of the short arm of chromosome 3 have been reported in a number of disorders featuring intellectual disability, such as schizophrenia (OMIM 181500; http://www.ncbi.nlm.nih.gov/omim); intellectual disability with language impairment and autistic features (OMIM 613670); Waardenburg syndrome, type 2A (OMIM 193510); Larsen syndrome (OMIM 150250); Zimmermann-Laband syndrome (OMIM 135500); spinocerebellar ataxia (OMIM 164500); and anemia Fanconi (OMIM 227646); in addition, abnormalities of these regions are also detected in cancer. 1 The subtelomeric aberrations affecting at least the band 3p25 produce 3p distal deletion syndrome (OMIM 613792). Deletions between 3p11 and 3p22.1 cause the proposed 3p proximal or interstitial deletion syndrome. 2
Kogame and Kudo reported the first case of interstitial deletion of the proximal short arm of chromosome 3; subsequently, 16 other cases of 3p interstitial deletions were described in the literature, with an additional 17 cases published in DECIPHER database (http://decipher.sanger.ac.uk/). Here, we add a new case of 3p interstitial deletion to the cases described heretofore. 2 –18
Case Summary
The 5-year-old male patient is the first child of nonconsanguineous parents, born after an uneventful pregnancy and delivery, with a delivery weight of 3500 g and an Apgar score of 9. At presentation, clinical investigation revealed a body weight of 17 kg (50th percentile), facial dysmorphism (square facies; broad forehead; short palpebral fissures, especially on the left eye; epicanthic folds; broad nasal bridge; short philtrum; small, malformed, low-set ears) (Figures 1A and B), hand and feet anomalies (small hands with short fingers; left fifth toe shorter and covered by the fourth toe); sacral sinus, hypochromic spot on the left shank; genital malformations (small penis, scrotal abnormalities, cryptorchidism); neurologic evaluation showed strabismus, bilateral profound hypoacusis, spastic diparesis, brisk osteo-tendinous reflexes, Babinski sign bilaterally, severe psychomotor development, severe speech delay (he says only 3 words, makes few simple orders), severe intellectual disability (intelligence quotient, IQ 34). The biological tests and electroencephalogram (EEG) were normal. Heart ultrasonography showed atrial septal defect, and cerebral X-ray computed tomography (CT) scan showed left cerebellum subarachnoidal cyst, large cisterna magna, moderate frontal atrophy, and bilateral sclerosis of inner ear bone chain.
The management of our patient included surgical intervention for congenital cardiac malformation, followed by physical therapy, ergotherapy, and cognitive stimulation.
Methods
Standard GTG-banding was performed on phytohemagglutinin-stimulated, cultured peripheric blood lymphocytes. Array-based comparative genomic hybridization (aCGH) was done on a 44 K Agilent platform, according to manufacturer’s protocol.
Results
Cytogenetic (GTG-banding) and molecular genetic (aCGH) investigations indicated the following karyotype: 46,XY,del(3) (p12p14) arr 3p12.3p14.1(65,317,864-77,902,945)x1 (build hg18) (Figures 1C and D).
Discussion
3p interstitial deletion syndrome is a rare condition, with 16 patients described in the literature since the first report, in addition to a similar number of cases enlisted in DECIPHER database. 2 –18 Deleted regions are variable in length and span from 3p11 to 3p22.1. A predominance of male patients is observed (Table 1). The characteristic disease-associated phenotype, that is, features present in a majority of patients, includes: psychomotor delay, facial dysmorphism (broad forehead, epicanthic folds, arched or cleft-palate/lip, short/broad nose, low-set malformed ears), clinodactyly, transverse palmar crease, hearing impairments, cardiac anomalies, genital anomalies (in boys only). All these characters were noted in our patient as well (Table 1). In addition, neurologic defects, brain, gastrointestinal, kidney, and urinary tract malformations—some present in our patient as well—were reported.
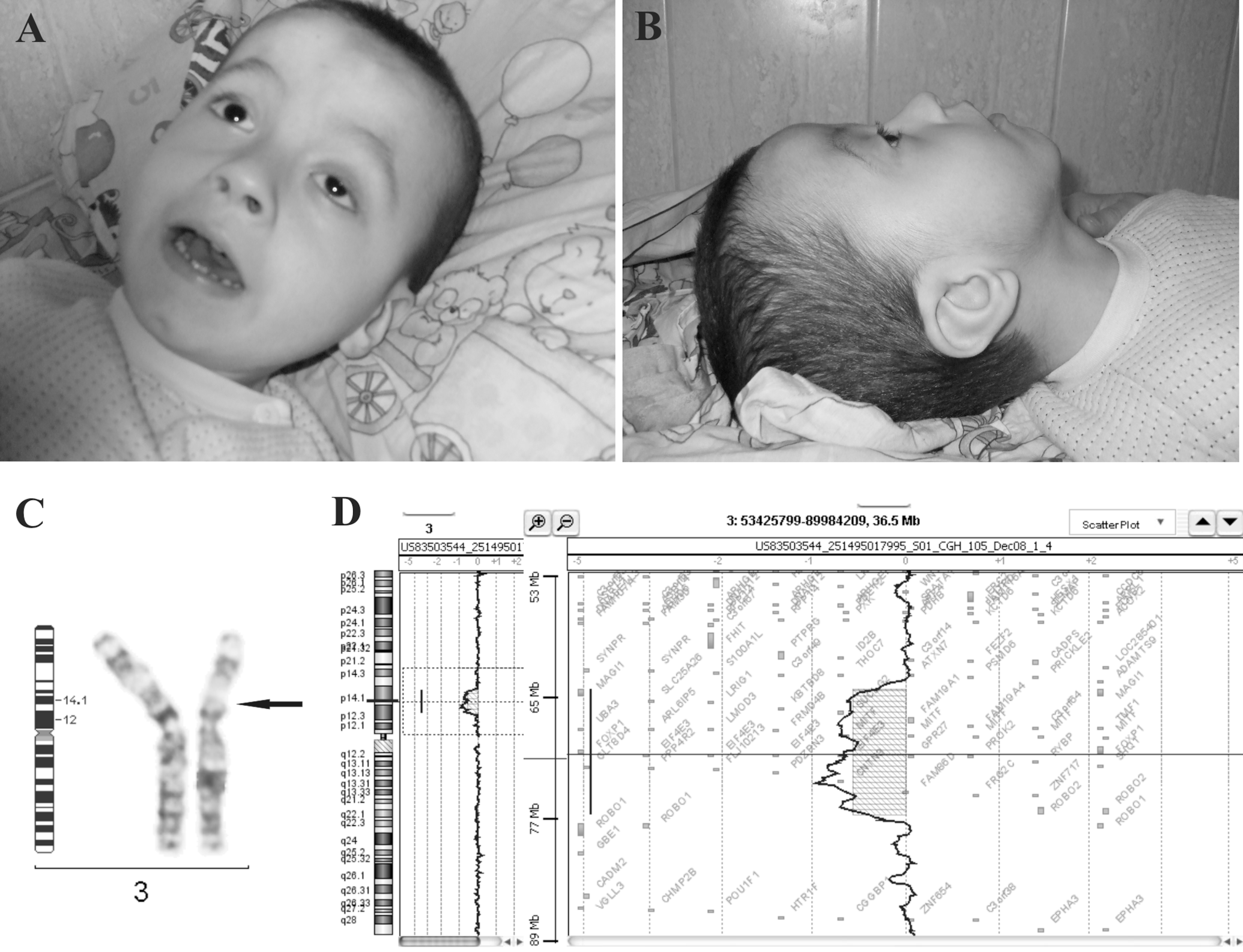
Facial dysmorphism of our patient: full face (A) and profile (B). (C) Partial GTG-banded karyotype showing an interstitial deletion on chromosome 3p. (D) aCGH profile showing a 12.5-Mb deletion on chromosome 3.
Cytogenetic Findings and Major Phenotypic Abnormalities Reported in Cases of 3p Interstitial Deletions
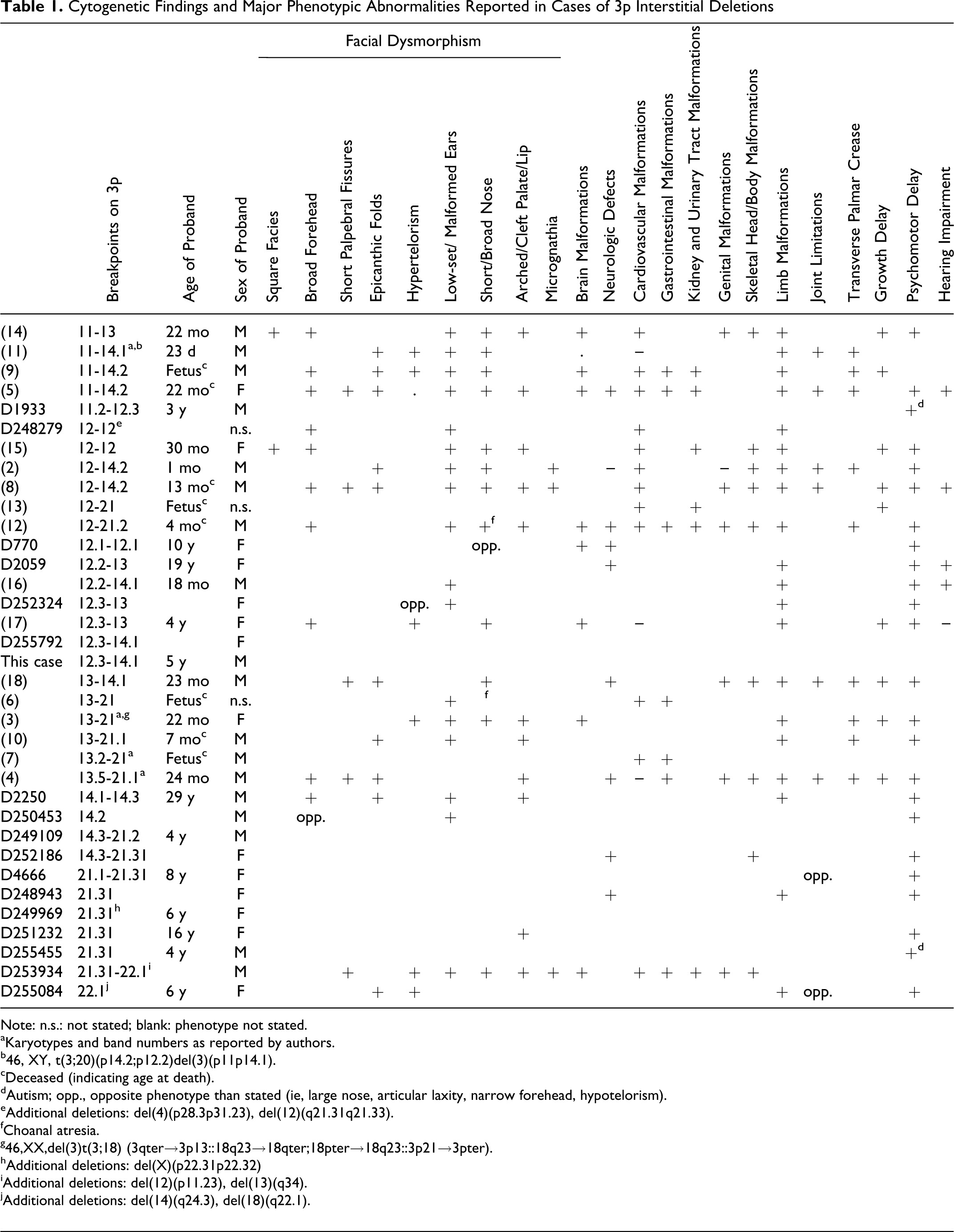
Note: n.s.: not stated; blank: phenotype not stated.
aKaryotypes and band numbers as reported by authors.
b46, XY, t(3;20)(p14.2;p12.2)del(3)(p11p14.1).
cDeceased (indicating age at death).
dAutism; opp., opposite phenotype than stated (ie, large nose, articular laxity, narrow forehead, hypotelorism).
eAdditional deletions: del(4)(p28.3p31.23), del(12)(q21.31q21.33).
fChoanal atresia.
g46,XX,del(3)t(3;18) (3qter→3p13::18q23→18qter;18pter→18q23::3p21→3pter).
hAdditional deletions: del(X)(p22.31p22.32)
iAdditional deletions: del(12)(p11.23), del(13)(q34).
jAdditional deletions: del(14)(q24.3), del(18)(q22.1).
The chromosomal region deleted in our case contains 31 protein coding ORFs (18 of which have known function), 36 pseudogenes, 5 miRNAs, and 13 hypothetical loci. 19 Among the genes with known functions, ROBO2, CNTN3, PROK2, PDZRN3, MITF, and FOXP1 are involved in cell migration and axonal navigation, cell differentiation, and/or development—particularly nervous system development. FOXP1 has been recently reported as mutated in autism spectrum disorders and nonsyndromic intellectual disability, 20 as well as in patients with intellectual disability and speech and language deficits. 21 In addition, many other genes act in cellular functions such as replication, repair, transcription, translation, ubiquitination, signaling, and cell adhesion, their disruption potentially affecting ontogenesis. Moreover, mitochondrial function—which was described to be responsible for neurodevelopmental disorders as well 22 —may be also impaired. The deletion of any of these genes or combination of genes may be causative of the observed phenotype. Surprisingly, multiple genes in these regions, including CNTN3, FOXP1, PDZRN3, PROK2, and ROBO2, have been reported as copy number variations (CNVs) in healthy subjects.
An additional aspect to be considered for the long-time management of our patient is related to the alterations in the copy number of the tumor and/or tumor suppressor genes located in this region—FOXP1 and LRIG1 in our case.
It may be concluded that the observed phenotype is the result of the deletion of multiple contiguous genes, rather than of a critical segment and the noted variability in the reported cases is finely modulated by the presence or absence of other neighboring genes. Our case and those described in the literature exhibit deletions spanning from 3p11 to 3p22.1. The cases listed in DECIPHER database show a more limited phenotype (which may be due, nevertheless, to the enlisting procedures); a deletion at 3p21.32-3p22.1, adjacent to the above-mentioned region, associates many phenotypic aspects, except for intellectual disability, while a smaller deletion concerning only 3p22.1 includes this disability. Therefore, further refinement of the critical region of this condition and of the characteristic phenotype is imperative.
Footnotes
Acknowledgments
This study makes use of data generated by the DECIPHER Consortium. A full list of centers that contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. The authors declare that those who carried out the original analysis and collection of the data published by DECIPHER Consortium bear no responsibility for the present analysis and interpretation. The authors thank Dr M. Perpelescu (National Institute of Genetics, Japan) and Dr M. L. Marton (University of Regensburg, Germany) for helpful discussions, Dr N. P. Carter (The Wellcome Trust Sanger Institute, United Kingdom) for great help in accessing DECIPHER database information, Dr S. Holden (DECIPHER principal investigator for Guy’s Hospital, London, United Kingdom) for access to patient data; and also to Mrs I. Borcan and Mrs M. Cristea for skillful technical assistance. In addition, the authors acknowledge the kind cooperation of the patient’s family.
Author Contributions
MB, KMR, and CD contributed to clinical description; ACTC, SMCP, and AA performed aCGH; ACTC prepared the manuscript, revised by AA and AL.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for the project was provided by the Wellcome Trust. This work was supported by the following grants: CNCSIS project 1203, PN 09.33.02.03, and PNII project 42-130.
Ethical Approval
The protocols for the collection of biological material, for experimental procedures, and information storage and disclosure comply with the Declaration of Helsinki, 1964, as revised by the 59th WMA General Assembly, Seoul, Korea, October 2008 and were approved by the ethical committees of all participating institutions. Informed consent for research and data publication was obtained from the legal guardian.