
Abstract

1. Maternal Prepregnancy Obesity and Maternal Vascular Malperfusion of the Placenta: a Study of >10,000 Recent Deliveries
1Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire
2Magee-Women's Research Institute, Pittsburgh, Pennsylvania
3University of Pittsburgh Graduate School of Public Health, Pittsburgh, Pennsylvania
Background
Obesity rates are increasing rapidly throughout much of the world. Maternal prepregnancy obesity increases the risk for numerous adverse pregnancy outcomes. However, only a few small studies have examined the placental pathology that develops in the context of the maternal prepregnancy obesity. This study examines the development of maternal vascular malperfusion with increasing maternal prepregnancy BMI.
Methods
We utilized our large perinatal and placental registry to identify all deliveries at our hospital between the years 2008 and 2012 for which a prepregnancy BMI was available (N = 26,395). For 10,674 (40.4%) of these cases, the placenta was sent for pathologic examination. Individual components of maternal vascular malperfusion (decidual vasculopathy, placental infarction, advanced villous maturation, intervillous fibrin deposition and perivillous fibrin deposition) were assessed, and a composite malperfusion score was generated. The data were adjusted for maternal age, race, smoking, education, parity and infant sex. Inverse weighting was used to correct for bias in the selection of placentas sent to pathology. The significance of the findings was only minimally changed by the inverse weighting.
Results
Increasing maternal prepregnancy BMI was significantly associated with both the presence of malperfusion (p = 0.0066) and a higher composite malperfusion score (p = 0.0232). Strikingly, maternal prepregnancy BMI correlated strongly with the presence of decidual vasculopathy (p < 0.0001). In particular, obesity more than doubled the likelihood of decidual vasculopathy, with an odds ratio of 1.98 (95% confidence interval 1.44-2.73) for obesity class I (BMI 30.0-34.9 kg/m2) and 2.43 (95% confidence interval 1.78-3.33) for obesity class II (BMI > 35.0 kg/m2).
Conclusion
These findings suggest that maternal prepregnancy obesity increases the risk of maternal vascular malperfusion, perhaps in part by significantly increasing the risk for decidual vasculopathy.
2. Clinicopathological Correlation of Large-for-Gestational Age Placenta in Pregnancies Complicated with Pregestational Diabetes
1Washington University School of Medicine, Saint Louis, Rhode Island
2Women & Infants Hospital of Rhode Island, Providence, Rhode Island
3Phoenix Perinatal Associates, Phoenix, Arizona
Background
Diabetic pregnancy is known to be associated with large infants, large placenta and increased adverse pregnancy outcome. Few studies have looked into the placental pathology and perinatal outcomes associated with large-for-gestational age (LGA) placentas in pregnancies complicated with pregestational diabetes (PreG DM). Current study aims to fill such a blank.
Methods
This was a retrospective cohort study. Women were identified through registration in the (state) Diabetes in Pregnancy Program from 2003 to 2011. Placental pathology report and related pregnancy information were extracted by chart review and stratified by placental weight percentile (AGA, 10–90th%tile; LGA, >90th%tile) in both type 1 (T1DM) and type 2 (T2DM) pregnancies. Categorical variables were compared by Chi-square1 or Fisher’s exact test2. Continuous variables were compared using t-test3. P < 0.05 is considered statistically significant.
Results
Total 293 gravida were identified with placental pathology reports, including 117 T1DM and 176 T2DM. In T1DM, there were 32 LGA placentas (32/117, 27.4%) and 63 (53.8%) AGA placentas. Postgrandial blood sugar level was significantly higher in the LGA group (P = 0.04). The prevalence of acute chorioamnionitis and meconium were both lower in LGA group (P = 0.02 and 0.04, respectively). Neonatal birth weight was significantly higher in the LGA group but the fetal-to-placental (f/p) weight ratio lower. In T2DM, there were 46 (26.7%) LGA and 90 (51.1%) AGA ones. The prevalence of villitis of unknown etiology is lower in the LGA group (2.1% vs 13.3%, P = 0.04). The gestational age at delivery was also earlier in the LGA group (37 vs 39 wks, P = 0.01). Same higher neonatal weight and lower f/p ratio were observed. In both groups, no difference of adverse pregnancy outcome was observed.
Conclusion
In our study population, about 25 to 30% pregnancies with preG DM produced LGA placentas. As expected, neonatal weight was significantly higher from the pregnancies associated with LGA placentas, but the f/p ratio lower, suggestive of lower placental efficiency. It is noted that some inflammatory conditions were seen less frequently seen in the LGA placentas. No significant differences of adverse outcome were seen between LGA and AGA group.
3. Pathologic Features of Revealed Acute Abruption – Timing is Everything
1Massachusetts General Hospital, Department of Pathology, Boston, Massachusetts
2Massachusetts General Hospital, Department of Obstetrics and Gynecology, Boston, Massachusetts
3Tufts University, Medford, Massachusetts
Background
The pathologic diagnosis of acute abruption has a poor correlation with the clinical diagnosis. This may be related to the timing of the abruption in relationship to delivery. Acute placental abruption can present with maternal bleeding per vagina (“revealed” abruption) or silently (“hidden” abruption). Determining the sequence of gross and histological findings in acute revealed placental abruption is the focus of this study. Our goal is to be able to “date” the onset of the abruption based on pathologic findings.
Methods
The XX Hospital pathology archives were searched over 15 years (2000-2015) for the clinical diagnosis of abruption. The 230 cases obtained were then screened for a revealed abruption with accurate bleeding times from onset of bleeding to delivery. After excluding multiple gestations, fragmented placentas, and complex bleeding times (more than one bleeding episode) the remaining 170 cases were re-examined by AC or DR for the presence (or absence) of features associated with acute abruption (retroplacental hematoma, intervillous hemorrhage, intravillous hemorrhage, trophoblastic necrosis, chorangiosis, acute villous edema, plasma cell deciduitis, acute chorioamnionitis, villitis of unknown etiology, hemosiderin, fetal normoblastemia, infarcts and other features of maternal vascular malperfusion). The pathological features were compared with recorded bleeding intervals: <30 minutes, 30 minutes – <60 minutes, 1–6 hours, >6–12 hours, >12–24 hours, 1to 3 days, > 3 days – 1 week.
Results
Most cases showed limited pathology even with bleeding times of up to one week. The most common finding was a retroplacental hemorrhage seen in up to 60% of cases within 3 hours of bleeding. Parenchymal indentation, intravillous hemorrhage and villous compression were present by 1 hour of bleeding time in >30% of cases. Most variables showed a weak linear relationship increasing over time except chorangiosis and placental infarct, which were inversely related over time. The only significant finding was that of plasma cell deciduitis which increased to ∼30% by one week from <1% at 30 minutes (R2 > .9).
Conclusion
Revealed acute abruptions are quite variable in gross and histologic features without a significant time course of progression of findings. It appears that 1 to 3 hours of bleeding is necessary to see most histopathologic features associated with abruption. Our study is limited in that we analyzed revealed abruptions. Perhaps the very fact that bleeding occurred decreased the retroplacental pressure and thus the histopathologic features. Revealed abruptions are difficult to time by gross and histopathologic findings. The most reliable features, intravillous hemorrhage and indentation, occur in >30% of cases by 1–3 hours of bleeding and plasma cell deciduitis which is a relatively late finding. Timing onset of acute revealed abruptions by gross and histopathologic features is not reliable.
4. Update on Stillbirth Classification and Comparing ReCoDe-R and Codac
1University of Ottawa, Ottawa, Ontario, Canada
2Trent University – Forensic Science Program, Ottawa, Ontario, Canada
Background
Stillbirth classifications provide the clinician with an identification guide of the possible cause for stillbirth, particularly in the presence of more than one risk factor. Minimizing the unexplained stillbirths is a fundamental goal of any classification system. Our aim is to evaluate stillbirth classification systems (ReCoDe-R and Codac) in establishing cause of stillbirth.
Methods
The study is a retrospective, non-interventional, observational audit for all cases of stillbirth of ≥22 weeks received at our institution between 2013 and 2015. Maternal charts, autopsy reports and related comprehensive ancillary tests findings were reviewed. Two expert reviewers analyzed all cases and classified causes according to ReCoDe-R and Codac classification systems.
Results
127 stillbirth autopsies were identified, of which 73 (57.5%) had an identified cause of death and 54 cases (42.5%) remained unexplained prior to the use of the classification systems. After using ReCoDe-R classification system, cause of death was identified in 83 cases (65.3%) and 44 (34.6%) cases remained unexplained. Codac classification system identified the causes of death in 91 cases (71.6%) and 36 (28.3%) cases that remained unexplained. The mean maternal age was 32.4 years (range 16–42). There were 31 mothers with an age of 35 years or greater (range 35 to 46 years). The mean BMI 24.8 (range 17–49). There were 4 mothers (4.2%) with recurrent stillbirth in which the cause of death was unexplained and the common risk factor for recurrence was BMI over 35. Among all autopsies, the most common cause of fetal demise is lethal congenital anomalies in 45 cases (35.4%) followed by placental complications in 24 cases (18.8%).
Conclusion
The use of stillbirth classification systems, particularly Codac system, was crucially helpful for identification of cause of death in stillbirths, which helps in further managing future pregnancies and direct resources.
5. Parenchymal Extrahepatic Non-reticuloendothelial Siderosis is not Specific to Hemochromatosis
Texas Children's Hospital, Houston, Texas
Background
Neonatal hemochromatosis (NH) is a severe liver disorder associated with extrahepatic siderosis sparing the reticuloendothelial system and leading to fetal death or neonatal liver failure. NH is considered a phenotypic expression of the liver abnormality regardless of etiology. Studies on the presence of extra hepatic, non-reticuloendothelial siderosis are limited with isolated case reports showing association with few congenital anomalies such as intestinal atresia, meconium ileus etc.
Methods
This is a retrospective study of autopsies at X hospital from 2010 to 2015. Autopsies of all stillbirths and early infant deaths (up to 8 weeks) were reviewed to include cases of liver failure as a significant cause of death. H&E and Prussian blue stained slides from selected organs such as liver, spleen, pancreas, thyroid, bronchial seromucinous glands, kidneys, adrenal glands and heart were reviewed in a blind fashion, for the presence of iron in both epithelial and stromal compartments. A semi quantitative analysis was performed using a three tier system: rare – single focus with less than 5 cells; focal – more than one focus; diffuse – easily identifiable in almost all fields. Clinical information was obtained from electronic medical records for clinicopathologic correlation.
Results
Ten cases of neonatal and post neonatal deaths due to liver failure were identified ranging from 7 to 42 days in age (mean 19.1 days) with male:female ratio of 4:3. Gestational age ranged from 35 to 39 completed weeks (mean 37.6 weeks). Infection was the most common cause of liver injury (4 herpes simplex virus, 2 adenovirus and 1 candida), others being congenital diaphragmatic hernia with hepatic necrosis (1), autosomal recessive polycystic kidneys with hepatic fibrosis (1) and abruptio placentae with hepatic necrosis (1). Seven out of 10 cases (70%) showed presence of extrahepatic non-reticuloendothelial iron of any extent. Thyroid showed follicular epithelial iron in 3/10 cases (30%) ranging from rare to diffuse. Pancreas showed islet and/or acinar iron deposits in 6/10 cases (60%) ranging from rare to focal. Kidneys showed tubular epithelial iron 4/10 cases (40%) ranging from focal to diffuse. Bronchial seromucinous glands showed epithelial iron in 2/10 cases (20%) ranging from rare to focal. Heart showed myocyte iron in 2/10 cases (20%) ranging from rare to focal and adrenal glands showed presence of rare cortical iron in 3/10 cases (30%). When present, iron was seen in at least 2 organs, with a combination of pancreas and kidney being the most frequent. All 10 cases showed diffuse hepatocellular iron accumulation with relatively less Kupffer cell iron. Spleen showed rare to focal iron deposition in these cases that was limited to hemorrhagic and congested areas.
Conclusion
Extrahepatic, non-reticuloendothelial iron deposition should not necessarily raise the suspicion for neonatal hemochromatosis, even in cases presenting with liver failure. It can be seen in a variety of tissue types, pancreas being the most common. Other causes of liver failure should be investigated as clinically and pathologically appropriate.
6. Refining the Diagnostic Criteria of Neuroendocrine Cell Hyperplasia of Infancy
1Stanford Hospital and Clinics, Stanford, California
2Seattle Children's Hospital, Seattle, Washington
Background
Neuroendocrine cell hyperplasia of infancy (NEHI) is a childhood diffuse lung disease whose etiology remains poorly understood. The current gold standard to diagnose NEHI is demonstration of increased bombesin-immunopositive (Bmb+) neuroendocrine cells (NECs) within a lung that is otherwise near normal histologically. The criteria for defining NEC excess are not well-established but experience dictates that at least 75% of non-cartilaginous airways have Bmb+ NECs, at least one airway has greater than 10% Bmb+ epithelium by area or cell number, and there are prominent clusters of NECs (neuroepithelial bodies-NEBs) around alveolar ducts. While other NEC markers are reported to be less sensitive than bombesin (Bmb) in demonstrating NEC increase, this has not been formally tested. Our objectives are: 1) To determine whether current criteria based on Bmb staining are sufficient to distinguish NEHI from other pediatric lung disorders, and whether other findings (NEB density, airway NEC clustering) further aid diagnosis 2) To determine whether more readily available NEC immunostains could be used in place of Bmb to diagnose NEHI.
Methods
Bmb, serotonin (Ser) and chromogranin (Chr) immunoreactivity are quantified in lung biopsies from 10 children with clinical presentation and imaging findings characteristic of NEHI, and compared to 10 age-matched cases biopsied for other lung disorders. The immunoreactivity of Bmb, Ser and Chr are compared quantitatively and qualitatively. Specific parameters measured are: 1) Number and type of airway with immunopositive NECs ((+)NECs) 2) Proportion of airway epithelial cells that are (+)NECs 3) Proportion of (+)NECs belonging to clusters 4) Density of NEBs.
Results
To date, we have fully quantified lung biopsies from 4 patients with clinical NEHI and 6 age-matched children with other lung disorders. We find that whilst Bmb is most sensitive in detecting NECs in both cases and controls, Ser and Chr show comparable staining patterns and can distinguish NEHI cases from non-NEHI cases.
Bmb, Ser and Chr staining show an increase in both the proportion of airway (+)NECs belonging to clusters (>50%) and NEB density (>0.15 cells/mm2) primarily in NEHI cases. Many non-NEHI cases, however, have at least 1 airway with more than 10% (+)NECs and all have at least 75% of airways with (+)NECs; thus these findings are not specific to NEHI. At least 3 airways with greater than 10% (+)NECs are observed primarily in NEHI cases.
A combination of 3 criteria: 1) At least 3 airways with greater than 10% (+)NECs 2) More than 50% (+)NECs in clusters 3) NEB density > 0.15/mm2, is sensitive and specific for NEHI, with all 4 NEHI cases fulfilling 2 or 3 of 3 criteria, and all 6 non-NEHI cases fulfilling 0 or 1 of 3 criteria.
Conclusion
More readily available immunostains for NECs, including Ser and Chr, are viable alternatives for the diagnosis of NEHI. NEC clustering and NEBs are increased in NEHI, findings that not only contribute to diagnostic specificity but also raise questions of how NEC clusters and NEBs relate to the biology of NEHI. Bmb is suggested to mark immature NECs, thus NEHI may represent not only NEC hyperplasia, but specifically a clustering of immature NECs. To this end, immunostaining for Mash1, a marker of NEC progenitors, may be insightful and is under way.
7. Response to Targeted Therapy with Mitogen-Activated Protein Kinase Pathway Inhibitors in Pediatric Disseminated Pilocytic Astrocytomas
1University of Texas Southwestern Medical Center, Dallas, Texas
2University of Texas Southwestern Medical Center and Children’s Health, Dallas, Texas
Background
Pilocytic astrocytomas (PAs) are the most common pediatric central nervous system tumor. Disseminated PAs without complete resection (CR) are a subgroup with poor prognosis and suboptimal response to conventional chemotherapy. Radiotherapy (RT) is the treatment of choice for PAs without CR; however, RT has significant long-term morbidity. Mitogen-activated protein kinase pathway (MAPK-P) activation associated with BRAF gene fusion or specific gene mutations has been shown to be the major molecular alteration in PAs. Here we present a series of PA patients with activated MAPK-P including response to targeted inhibitor therapy.
Methods
Pediatric patients with progressive disseminated PAs who received treatment at our center in the past year (2015–2016) were retrospectively identified. As part of clinical care, the patients’ formalin-fixed paraffin-embedded tumors were analyzed by next-generation sequencing panels (including BRAF, KRAS, NRAS, and PIK3CA) using an in-house clinical test (25 Genes) or Foundation Medicine testing (315 Genes). Identified mutations were used to guide targeted therapy with either dabrafenib (BRAF inhibitor) or dabrafenib combined with trametinib (MEK inhibitor). Response to treatment was assessed by clinical status and imaging.
Results
Five patients (3 females, mean age = 8.4 years) with disseminated PAs were identified. Three of them had spinal leptomeningeal dissemination. Four patients had a MAPK-P activating variant: BRAF V600E (n = 2), BRAF V600D (n = 1), KRAS G60_Q61ins7 (n = 1). Two patients (BRAF V600E, V600D) with progressive disease were not suitable candidates for RT and were treated with dabrafenib. One patient (BRAF V600E) responded with partial disease resolution and has been clinically stable for 11 months. One patient (BRAF V600D) had initial improvement of leptomeningeal tumor burden, but progression of the primary tumor. After 6 months, trametinib was combined with dabrafenib; subsequently, the tumor burden has decreased and the patient has had 5 months of stable disease.
Conclusion
MAPK-P activation is well-known in pediatric PAs; however, there are few reports on the therapeutic implications of pathway inhibition. In this study, the majority of cases have non-fusion activating events (4 of 5 cases) with frequent genetic variants novel to PAs: BRAF V600D and KRAS G60_Q61ins7. The patients treated with MAPK-P inhibition have had clinical response and minimal toxicity (skin rash). Patients with progressive, disseminated PAs may benefit from routine mutation analysis and targeted therapy.
8. Human Parechovirus 3 is the Leading Picornavirus Associated with CNS-infections in Children
Childrens Mercy Hospital, Kansas City, Missouri
Background
Enterovirus (EV) and human parechovirus (HPeV) are small non-enveloped RNA viruses in the Picornaviridae family. Together these viruses are the leading cause of central nervous system (CNS) infections in children during summer-fall months each year. Although a total of 16 HPeV and 118 EV types are currently known to circulate only few types are associated with CNS infections. For example, HPeV3 is a recently emerged and predominant HPeV type associated with CNS infection. However, knowledge and awareness of the HPeV CNS-infection is lacking due to limited availability of HPeV specific testing in children’s hospital. We recently reported HPeV3 CNS-infections in children during 2007 and 2009. The goal of this study is to determine the prevalence of various EV and HPeV types causing CNS-infections in children during 2007 and 2009.
Methods
All EV positive CSF specimens from 2007 and 2009 were forwarded to CDC for genotyping by amplification and nucleotide sequencing of the VP1 viral capsid protein gene. The EV type data was compared with previously typed HPeV positive CSF specimens to determine prevalence of both viruses.
Results
HPeV was detected in a total of 17% (54/320) in 2007 and 18% (66/362) in 2009 CSF specimens tested. EV was detected in a total of 20% (86/421) in 2007 and 13% (55/427) in 2009 CSF specimens. HPeV3 was the predominant HPeV type among the successfully typed HPeV positive specimens available for sequencing in 2007 (97%; 48/49) and 2009 (100%; 51/51). Unlike HPeV, 19 different EV types were detected in 130 EV-positive specimens, the top 5 types were echovirus 18(22%; n = 28), coxsackievirus B4 (15.3%; n = 20), coxsackievirus A9(10.7%; n = 14), echovirus 5 (7.7%; n = 10) and echovirus 7(6.1%; n = 8). Some EV types were prevalent in both years (echovirus 18, coxsackievirus A9) while others were restricted to either the 2007 (coxsackievirus B4, echovirus 5) or 2009 (echovirus 7) season. Overall, HPeV 3 (n = 99) was determined to be the leading cause of CNS infections in children during both 2007 and 2009 followed distantly by echovirus 18 (n = 28).
Conclusion
We document that HPeV3 is the leading Picornavirus associated with CNS-infections in children. Our data provides strong evidence that HPeV CSF-testing will significantly increase the diagnostic yield and recommend it to be included in test menu for CSF-testing in children especially during the summer-fall season of each year
9. Evaluation of the Utility of Bone Marrow Morphology and Ancillary Studies in Patients under Surveillance for MDS/AML
Seattle Children’s Hospital, Seattle, Washington
Background
Inherited bone marrow failure syndromes such as Fanconi anemia (FA), dyskeratosis congenita (DKC), and Shwachman-Diamond syndrome (SDS) have a propensity to develop into myelodysplastic syndrome (MDS) and/or acute myeloid leukemia (AML). At our institution, patients with marrow failure syndromes are screened at least annually with bone marrow examinations. These marrows are sent for flow cytometry, cytogenetics (karyotype), and MDS fluorescence in situ hybridization (FISH), the latter panel detecting deletion 5q/monosomy 5, monosomy 7/deletion 7q, trisomy 8, and deletion 20q. In the current study, we evaluate the utility of these tests.
Methods
Our pathology database was queried for cases in which the MDS FISH panel was performed between January 2014 and May 2016. The results of these surveillance examinations were reviewed. Patient data including history (bone marrow failure syndrome), sex, and age at time of marrow were also documented. Patients status post transplant were excluded.
Results
A total of 132 patients (M:F = 64:68) and 188 cases were identified as surveillance marrows. Diagnoses included neutropenia (n = 23), aplastic anemia (AA, n = 22), SBD (n = 12), FA (n = 11), DKC (n = 7), Diamond-Blackfan anemia (DBA, n = 6), chronic immune thrombocytopenic purpura (ITP, n = 6), other thrombocytopenia (n = 6), pancytopenia (n = 8), bicytopenia (n = 7), known MDS (n = 4), other anemia (n = 4), essential thrombocythemia (ET, n = 2), and other (n = 14). Median age at time of bone marrow was 8.0 years. Of all 188 cases, 134 (71%) demonstrated normal morphology, 39 demonstrated single lineage dysplasia, 13 had bilineage dysplasia, and 2 had trilineage dysplasia. Of the 142 cases with flow cytometry performed, 135 (95%) had no abnormalities, 3 showed paroxysmal nocturnal hemoglobinuria clones, 2 showed increased and abnormal blasts, and 2 showed small abnormal B cell populations (<1% total cells) of uncertain significance. No flow cytometry cases detected abnormal myeloid antigen expression except for the two with increased blasts. Of the 184 cases with cytogenetics performed, 164 (87%) had normal karyotypes, 4 had constitutional abnormalities, 3 had multiple abnormal clones, 7 had deletion 20q, 3 had monosomy 7/deletion 7q, 2 had isochromosome 7q, and 1 had trisomy 8. Of the 181 cases with MDS FISH performed, 168 (93%) had no abnormalities, 5 had monosomy 7/deletion 7q, 7 had deletion 20q, and 1 had isochromosome 7q. All cases with abnormal MDS FISH also had an abnormal karyotype; furthermore karyotype detected 3 additional abnormal cases. Of the 16 cases with acquired abnormal karyotypes, 10 also showed morphologic dysplasia; two of these cases with abnormal karyotypes and morphologic dysplasia also had increased blasts detected by flow cytometry.
Conclusion
We demonstrate that the combination of morphologic evaluation and cytogenetics (karyotype) without flow cytometry and/or MDS FISH is sufficient to detect the abnormalities for these indications. Rapid review with selection of those with potential benefit by flow is necessary to curtail excess use. Unnecessary testing can be reduced without decreasing the sensitivity of detecting myelodysplasia.
10. Prevalence of De-novo Non-alcoholic Fatty Liver Disease in Children Undergoing Liver Transplant for Other Conditions
1Baylor College of Medicine, Houston, Texas
2Texas Children's Hospital, Houston, Texas
Background
The development of de novo non-alcoholic fatty liver disease (NAFLD) or steatohepatitis (NASH) following orthotopic liver transplantation (OLT) is a well described complication in adult patients who have undergone OLT for unrelated causes. However, the prevalence of this condition in the pediatric population after OLT is largely unknown. Given the increasing prevalence of metabolic risk factors in the pediatric population, the known metabolic risks posed by immunosuppressive regimens and the increasing long term survival of pediatric patients following OLT, it is likely that this is not an uncommon occurrence. It is the aim of our study to evaluate the incidence and prevalence of NAFLD/NASH in a pediatric cohort after OLT and to identify potential risk factors leading to its development in this population. This can in turn lead to improved therapeutic and follow up strategies for these patients.
Methods
Institutional pathology files were searched for the co-occurrence of terms “liver” and “transplant or allograft” in the final diagnosis. Cases were then filtered to include all post-transplant liver biopsies from 2002 to 2015. Pathologic data was tabulated and clinical information was obtained from electronic medical records. Significant steatosis is defined as >5%. A subset of data is presented as abstract.
Results
A total of 250 biopsies from 69 patients (M:F 1.4:1) were identified with age at biopsy ranging from 5 months to 22 years (median 7.3 years), transplanted for various reasons, biliary atresia being the the most common (24/69, 35%). Post-transplant interval at biopsy varied from 8 days to 20 years (mean 2.2 years). Microscopically, 46 biopsies (18%) belonging to 27 patients (39%) showed the presence of macrovesicular steatosis, of which 26 biopsies (10.4%) belonging to 15 patients (21.7%) had greater than 5% steatosis. Of these, the majority (11 pts, 73.3%) showed grade 1 steatosis, with 1 pt (6.6%) showing grade 2 and 3 pts (20%) showing grade 3. No patient showed steatohepatitis. Interestingly, all 3 patients with grade 3 steatosis had cystic fibrosis; one of which showed similar degree of steatosis in the native liver. Among the non-CF patients, biopsies with >5% steatosis were significantly more associated with acute cellular rejection (88.5%) when compared to those with no/minimal steatosis (62.5%), p 0.008 by Fisher's exact test and also higher incidence of recent steroid use (65.4% vs 28.6%) p 0.0007 by Fisher's exact test. Earliest interval for the development of significant steatosis was 2 months post-transplant (median 2.3 years). There was no significant difference in the BMI of patients with and without significant steatosis.
Conclusion
Significant post-transplant macrovesicular steatosis is seen in a minority of post-transplant children with cystic fibrosis being the only condition associated with severe grade 3 steatosis. In non-CF patients, episodes of acute cellular rejection and/or steroid use seem to be the risk factors for development of steatosis.
11. Hepatocellular Malignant Neoplasm-NOS: A Clinicopathologic Study of 11 Cases from a Single Institution
1Children's Hospital Los Angeles, Los Angeles, California
2Texas Children's Cancer Center, Houston, Texas
Background
Hepatocellular malignant neoplasm-NOS (HMN-NOS) is a new provisional entity describing a small subset of malignant hepatocellular tumors, which typically occur in older children/young adolescents and have histological features neither typical hepatoblastoma (HB) or hepatocellular carcinoma (HCC). The term “transitional liver cell tumor” was initially proposed for this group of tumors. Controversy exists as to whether these tumors are HBs with chemotherapy induced HCC-like histology, clonally progressed HBs, or a distinct group from both HB and HCC.
Methods
To better characterize this entity, we retrospectively evaluated clinical and pathological features of patients with histological features of neither typical HB nor HCC.
Results
Eleven patients (10 males and 1 female) were identified at our institution from Jan. 2000 to Mar. 2016. The median age and serum alpha fetoprotein at diagnosis was 7 years (range, 4 to 15 years) and 131,000 ng/ml (range, 617 to1.28 million ng/ml), respectively. The majority of patients presented with right upper quadrant abdominal pain and three patients presented with acute abdomen due to tumor rupture. All tumors (8 in the right lobe, 1 in the left and 2 involving both lobes) were large with at least two separate masses. One patient was PRETEXT stage II, 7 stage III and 3 stage IV. Three patients had lung metastases. Two patients with tumor rupture underwent upfront gross total resections. After neoadjuvant therapy, seven patients had complete surgical resections including three primary liver transplantations and two patients had microscopic residual disease. Extensive vascular invasion was seen in 10 of 11 tumors. All patients received chemotherapy, the majority including cisplatin, vincristine, 5-flourouracil and doxorubicin. Other agents included ifosfamide, interferon, irinotecan, oxaliplatin, sorafenib, and gemcitabine. Two patients had tumor recurrence and one patient underwent a liver transplantation following local recurrence. All patients had either a biopsy or resection specimen before chemotherapy. The original pathology diagnoses were: HB in 7 patients, HCC in 2 and HMN-NOS in 2. After chemotherapy, pathology diagnosis was changed from HB to HMN-NOS in one case and from HCC to HB in another case. Our pathology review of pre-chemotherapy specimens showed that ten of 11 cases had fetal/embryonal HB histology along with focal HCC-like histology and one with HB histology only. Post-chemotherapy resection specimens showed predominant HCC-like histology with focal HB histology in 7 cases, predominant HB in 1 and complete tumor necrosis in 1. A panel of beta-catenin, glypican 3 and SALL4 immunostaining showed that all tumors had a mixed HB/HCC immunophenotype that corresponded to their histology features. At a median follow-up 6.1 years (range 4 months to 11.8 years), all patients were alive, 10 in complete remission, and 1 with persistent disease.
Conclusion
HMN-NOS have mixed HB/HCC histology and an aggressive clinical phenotype but patients have a high survival rate with aggressive treatment. Our data suggest that HMN-NOS may be clonally progressed HBs. Further studies evaluating the molecular basis for this group of tumors are needed.
12. DUX4 Immunohistochemistry is a Distinctly Sensitive and Specific Marker for CIC-DUX4 Fusion-Positive Round Cell Tumor
1University of Colorado School of Medicine, Aurora, Colorado
2Children's Hospital Colorado, Aurora, Colorado
3Seattle Children's Hospital, Seattle, Washington
4Boston Children's Hospital, Boston, Massachusetts
Background
The histologic differential diagnosis of pediatric and adult round cell tumors is vast and includes the recently recognized entity CIC-DUX4 fusion-positive round cell tumor. The diagnosis of CIC-DUX4 tumor can be suggested by light microscopic and immunohistochemical features. Currently, definitive diagnosis requires ancillary genetic testing such as conventional karyotyping, fluorescence in situ hybridization, or molecular methods; however, there remain limitations inherent to each of these modalities. We sought to determine whether DUX4 immunohistochemistry can reliably distinguish CIC-DUX4 tumors from potential histologic mimics.
Methods
We reviewed round cell tumors and undifferentiated sarcomas from pediatric and young adult patients for which CIC-DUX4 fusion-positive round cell tumor might be considered in the differential diagnosis. Cases included 5 CIC-DUX tumors, 5 Ewing sarcomas, 1 Ewing-like sarcoma, 2 alveolar rhabdomyosarcomas, 2 embryonal rhabdomyosarcomas, 2 synovial sarcomas, 2 desmoplastic small round cell tumors, 3 malignant rhabdoid tumors, 1 neuroblastoma, 1 clear cell sarcoma, 1 B-cell acute lymphoblastic leukemia, and 3 basal cell carcinomas. Paraffin-embedded sections were stained by standard immunohistochemical (IHC) methods with a commercially-available monoclonal antibody raised against the C-terminus of the DUX4 fusion protein (Thermo Fisher Scientific, Waltham, MA). All CIC-DUX4 fusion-positive tumors were confirmed by cytogenetic and/or molecular methods.
Results
DUX4 immunohistochemistry exhibited diffuse, crisp, strong nuclear staining in all CIC-DUX4 fusion-positive round cell tumors (5/5; 100% sensitivity) and negative nuclear staining in all of the other tested round cell tumors (0/23; 100% specificity).
Conclusion
In our study, DUX4 fusion protein-based immunohistochemistry was 100% specific and sensitive for the identification of CIC-DUX4 fusion-positive round cell tumor. The method is readily available as part of routine immunohistochemistry, and thus we highly recommend its use in the diagnostic workup of round cell tumors and undifferentiated sarcomas in everyday practice.
13. Utility of Phox2B Immunohistochemical Stain in Pediatric Tumors
Children's Hospital Los Angeles, Department of Pathology and Laboratory Medicine, Los Angeles, California
Background
Recently Phox2B has emerged as a highly sensitive and specific marker for autonomic nervous system (ANS) tumors, i.e. peripheral neuroblastic tumors (pNTs) of neural crest origin, with suggestion that its expression may be the only factor to support the diagnosis of this group of tumors. However, its utility for this diagnostic role has not fully been assessed. We thus compared expression of Phox2b immunohistochemically along with another neural crest marker, tyrosine hydroxylase (TH), in pediatric neural crest tumors and non-neural crest tumors.
Methods
Phox2b immunohistochemical (IHC) stains were performed on tumors in 4 different categories—(1) neural crest tumors with ANS neuronal differentiation (61 pNTs): 11 neuroblastoma undifferentiated (NB-UD), 10 NB poorly-differentiated (NB-PD), 10 NB differentiating (NB-D), 10 ganglioneuroblastoma intermixed (GNB-IM), 10 GNB nodular (GNB-N), and 10 ganglioneuroma maturing/mature (GN-M); (2) neural crest tumors with neuroendocrine differentiation (20 cases): paragangliomas/pheochromocytomas (PG/PCC); (3) neural crest tumors without neuronal/neuroendocrine differentiation (24 cases): 4 schwannomas, 9 melanomas, 8 melanocytic nevi, and 3 melanocytic neuroectodermal tumors of infancy; and (4) non-neural crest tumors with or without neuronal differentiation (38 cases): 5 Ewing sarcomas/PNETs, 4 olfactory neuroblastomas, 1 retinoblastoma, 8 rhabdomyosarcomas, 2 alveolar soft part sarcomas, 2 desmoplastic small round cell tumors, 2 clear cell sarcomas, 3 synovial sarcomas, 2 Wilms tumors, 5 malignant rhabdoid tumors, and 4 lymphoblastic lymphomas. Additionally, TH IHC was performed on tumors of categories (1) and (2). We also reviewed Phox2b and TH IHC expression in one composite rhabdomyosarcoma (rhabdomyoblastic and neuroblastic differentiation) and 8 normal adrenal medullas.
Results
All pNTs diffusely expressed Phox2b: uniformly strong nuclear positivity for neuroblasts in NB-UD, NB-PD, and NB-D, and positive nuclear staining with variable intensity for ganglion cells in GNB-IM, GNB-N, and GN-M. While neuroblasts in NB-PD and NB-D, as well as ganglion cells in GNB-IM, GNB-N, and GN-M, showed diffuse and strong cytoplasmic TH (n = 49/50), 7 of 11 NB-UDs did not express this marker. The neuroblastic nodule in one GNB-N was also negative for TH. All PG/PCC (n = 20/20) showed diffuse but variable nuclear staining for Phox2b and diffuse and strong cytoplasmic staining for TH. Normal adrenal medulla also stained diffusely with Phox2b and TH. The composite rhabdomyosarcoma expressed Phox2b and TH only in the neuroblastic component. All other tumors were negative for Phox2b.
Conclusion
Expression of Phox2B was limited and specific to the neural crest tumors with neuronal/neuroendocrine differentiation. Diffuse and strong Phox2b expression was especially useful for identifying NB-UD, which often lacked TH expression. In contrast, neural crest tumors without neuronal/neuroendocrine differentiation and non-neural crest tumors, including major pediatric small round cell tumors, were negative for Phox2B. Our study also suggested that the composite rhabdomyosarcoma represents a neural crest tumor with diverse differentiation.
14. Role of HBME-1, Galectin-3 and CD56 in the Diagnosis of Follicular Variant of Papillary Thyroid Carcinoma in Children
1University of Missouri, Kansas City- Truman Medical Center, Kansas City, Missouri
2Children's Mercy Hospital, Kansas City, Missouri
3Children's Healthcare of Atlanta, Atlanta, Georgia
4Nationwide Children's Hospital, Columbus, Ohio
Background
The incidence of thyroid cancer has been increasing in children in recent years. In children, the leading three histologic types of thyroid cancer by frequency of occurrence are papillary thyroid cancer (PTC), follicular variant of papillary thyroid carcinoma (FVPTC) and follicular carcinoma (FC). The diagnosis of FVPTC could in several instances be fraught with interobserver disagreement and intraobserver disagreement. The diagnostic criteria of follicle formation and optical clearing of nuclei, is not unique to FVPTC, and atypical benign follicular nodules may morphologically mimic FVPTC. Several immunohistochemical (IHC) markers like HBME-1, galectin-3, CD56 and others have been studied in adult thyroid cancers and have been noted to be helpful in the diagnosis of papillary carcinoma and its variants. We undertook a study of FVPTC tumors and benign follicular nodules diagnosed in children and compared their immunohistochemical expression of a limited panel of antibodies.
Methods
Institutional review board approval was obtained for this HIPAA-compliant study. The requirement for written informed consent was waived. We retrospectively selected 66 cases (32 FVPTC and 34 benign follicular nodules) from three tertiary care hospitals for children (age range 1–17 years). The benign follicular nodules included 23 follicular adenomas (FA) and 11 adenomatoid nodules (AN). Formalin-fixed paraffin embeded tissue from representative tumor blocks was submitted for Hematoxylin and Eosin stain (H&E stain) and IHC for HBME-1, galectin-3 and CD56. The slides were evaluated by observers who were blinded to the institutional diagnoses. The extent of IHC expression in the lesional cells of the tumor nodules was scored as absent, rare (<25% of the cells), focal (25–75% of cells) and diffuse (>75% of cells). For the analysis of IHC stains, absent and rare staining pattern were combined to denote ‘negative’; whereas, focal and diffuse staining pattern were combined to denote ‘positive’. Further, the diagnostic categories were grouped into two categories FVPTC and benign follicular nodules (FA and AN).
Results
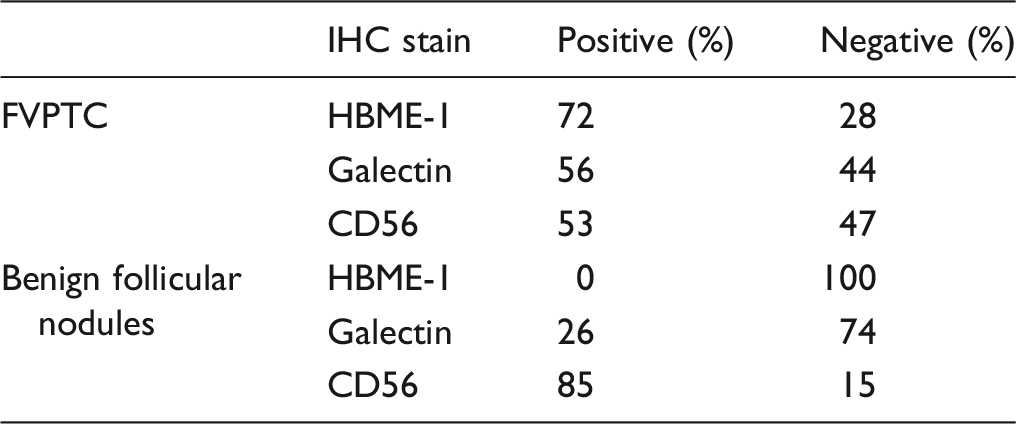
Conclusion
The study revealed that there is significant interobserver disagreement in the diagnosis of FVPTC. Since all benign follicular nodules were negative for HBME-1 and mostly positive for CD56, we conclude that a histological diagnosis of BFN can be supported by the use of these antibodies. Our study also noted that the diagnosis of FVPTC is strengthened if there is diffuse expression of HBME-1 in the lesion. Thus, using HBME-1 and CD56 IHC may help to reduce the interobserver disagreement in the diagnosis of FVPTC.
15. IGF1 and bFGF Signaling via Akt Maintains Tumor Stem Cell Survival in Large/Giant Congenital Nevi
1Departments of Pathology, CHP, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania
2Division of Plastic and Reconstructive Surgery, North Shore University, Northbrook, Illinois
Background
Tumor recurrence and malignant transformation from large/giant congenital melanocytic nevi (L/GCMN) and Neurocutaneous melanocytosis (NCM) continues to be a major concern. Surgical interventions do not reduce melanoma risk. Tumor stem cells are thought to be the source of tumor repopulation and recurrence. We and others have recently identified clonogenic tumor initiating cells (TICs) in L/GCMN and NCM. TICs carry the same oncogenic mutation as the parent nevi and grow as ‘nevosphere’ colonies. However, factors supporting the survival and maintenance of TICs have not yet been identified. Our aim is to define the factor(s) supporting tumor stem cell survival in L/GCMN.
Methods
Nevocytes were isolated from 3 L/GCMN skin lesions: two carrying NRASQ61K and one BRAFV600E. Colony formation assays on a collagen-laminin containing gel matrix were performed in the presence or absence of a defined set of growth factors. Clonogenicity was assessed using media with and without IGF1 or bFGF and after treatment with sub-lethal doses of MEK inhibitor (MEK162) or PI3K/mToR inhibitor (GSK2126458). Cell viability assay (MTT assay using the Cell Titer 96 Non-Radioactive Cell Proliferation Assay kit® – Promega, USA) and phosphorylation of Akt and ERK1/2 (by immunoblotting) were performed in cells treated with increasing doses of IGF1 or bFGF.
Results
The total number of colonies decreased by approximately 50–55% in all cell lines when cultured in IGF1 or bFGF – free media as compared to media containing IGF1 or bFGF. Conversely, adding back IGF1 and/or bFGF to culture medium rescued colony formation efficiency to the same extent. Dose dependent increase in phosphorylated Akt and ERK 1/2 was observed upon treatment of nevocytes with recombinant IGF1 and bFGF. Inhibiting ERK1/2 by 1 μM MEK162 appeared to have no significant impact on clonogenicity whereas Akt inhibition by 0.5 μM GSK2126458 completely inhibited colony formation.
Conclusion
Both IGF1 and bFGF signaling is important for the growth and survival of TICs from L/GCMN and NCM in vitro. IGF1 and bFGF signaling intermediates could be potential therapeutic targets towards elimination of tumor stem cells in L/GCMN and NCM.
16. Serotonin Expression in Large/Giant Congenital Melanocytic Nevus (L/GCMN)
1Departments of Pathology, CHP/University of Pittsburgh, Pittsburgh, Pennsylvania
2Division of Plastic and Reconstructive Surgery, NorthShore University HealthSystem, Northbrook, Illinois
Background
Pruritus in the melanocytic lesion is a frequent symptom of patients with L/GCMN. Some of these patients have severe pruritus and do not respond to anti-histaminic medication, but there are reports of improvement with serotonin receptor-type3 antagonist. We have demonstrated an increased number of mast cells in skin from patients with large/giant CMN, but the expression of serotonin in L/GCMN has not be systematically studied. Our aim was to examine the expression of serotonin in nevus from patients with L/GCMN with NRAS or BRAF mutation.
Methods
Prospectively collected melanocytic lesions from patients with L/GCMN, with mutations in codon 61 of NRAS or in codon 600 of BRAF, were selected from “The XXX Bio-repository”. Immunohistochemistry for Serotonin (SIGMA, S5545; 1:5.000 dilution) and for tryptase (DAKO, Clone AA1; 1:200 dilution) were performed on the Ventana® Benchmark Ultra, with adequate controls.
Results
We studied 16 nevi; 10 with NRAS codon 61 mutation (8 giant and 2 large) and 6 with BRAF V600E mutation (3 giant and 3 large). In all nevi the expression of serotonin was strong in cells that also express tryptase (mast cells). Nevus cells of 13/16 (81%) lesions expressed serotonin (6/6 BRAF and 7/10 NRAS mutated nevus). The expression of serotonin in nevus cells was strong in the superficial cell layer or nests in 4/6 BRAF and in 4/10 NRAS mutated nevus. Nevus cells in deeper layers (deep reticular dermis) of the nevus do not express serotonin. There was no association between serotonin expression and nevus size, location, type of mutation or presence of neurocutaneous melanocytosis.
Conclusion
Mast cells and superficial nevus cells of some L/GCMN express serotonin. The presence of serotonin in L/GCMN could be one of the factors responsible for the pruritus in these patients and could explain their response to the serotonin receptor antagonist.
17. Exploring the Inflammatory Profile of Cardiac Allograft Vasculopathy in Pediatric Transplant Recipients
University of Chicago, Chicago, Illinois
Background
Cardiac allograft vasculopathy (CAV) remains a major cause of late graft loss in pediatric heart transplant patients. Immune and nonimmune processes contribute to the diffuse and progressive, concentric intimal thickening of proximal coronary arteries. Repeated episodes of acute cellular (ACR) and antibody-mediated rejection (AMR) are variably associated with increased risk for CAV and may be augmented during adolescence due to enhanced immune system function and effects of pre-existing graft coronary disease. Characterization of the inflammatory profiles of epicardial vessel lesions in CAV may offer insight into the contributing immune responses in the pediatric cardiac transplant population.
Methods
From seven cases of CAV (six autopsies and one cardiac allograft explant) in patients who underwent cardiac transplantation at age 21 or younger (mean age at transplantation 12 ± 7.4 years), formalin-fixed, paraffin-embedded (FFPE) sections of left ventricular tissue and epicardial coronary vessels were examined with H&E stains. Intimal and medial wall thicknesses of the largest vessel in each section were measured. Additional sections were stained for CD3, CD68, and CD20 by immunohistochemistry (IHC) for identification and localization of T-cells, intravascular activated mononuclear cells, and B-cells, respectively. Cells of each type were quantified in areas of greatest prevalence in the large epicardial vessels, myocardial interstitial space, and smaller intracardiac vessels and reported as low (<20 cells per high power field [HPF]), moderate (20-100 cells per HPF), or high (>100 cells per HPF). C4d IHC stains were also performed to classify immunopathologic AMR.
Results
CD3- and CD68-positive cells were present in the intimal layer of large epicardial vessels in moderate to high quantities in six out of the seven cases with sparing of the medial layer. Intimal wall thickness tended to be increased relative to that of the medial layer in cases with diffuse C4d-positive stains consistent with immunopathologic AMR. CD20-positive cells and plasma cells were generally absent from CAV lesions.
Conclusion
In pediatric cardiac allograft patients with CAV, a distinct T-cell and macrophage infiltrate localizes to the intimal layer of large epicardial arteries in a diffuse, concentric pattern without medial layer involvement. These findings are consistent with the paradigm of endothelial activation as a key step in pathogenesis of CAV as described in adult patients. Further work is ongoing to increase the number of patients and characterize the effects of ACR and AMR on the development of CAV in the pediatric cardiac transplant population.
18. Esophageal Pathology in Children with IBD: Lymphocytic Esophagitis Favors a Diagnosis of Crohn's Disease
Hospital for Sick Children, Toronto, Ontario, Canada
Background
Initial evaluation of pediatric patients with a possible diagnosis of inflammatory bowel disease (IBD) includes upper endoscopy. Some studies suggest that the presence of lymphocytic esophagitis (LE) favors Crohn’s disease (CD) in the pediatric age population. However, this is not a consistent finding and further work is required to confirm this association. The aim of the present study was to investigate the association of LE with IBD and its subtypes in a pediatric population.
Methods
Pre-treatment esophageal biopsies from children with (subsequent) clinically and histologically confirmed inflammatory bowel disease were studied. There were a total of 124 IBD patients (71 with CD and 53 with ulcerative colitis (UC)). A group of non-IBD patients (n = 21), excluding those with primary esophageal pathology, was used as controls. All biopsies were reviewed in a blinded fashion and assessed for LE, eosinophilic esophagitis (EoE) and granulomas. LE and EoE were defined using previously described criteria. Tabulated data were compared using Fisher’s exact test.
Results

Conclusion
LE was significantly more common in IBD patients than in non-IBD controls. Within the IBD group, the presence of LE was significantly associated with CD. Our data supports previous studies, and confirms that the finding of LE strongly favors the diagnosis of CD in pediatric IBD patients.
19. The Incidence, Clinicopathologic Spectrum and Natural History of Pediatric Syndromic and Nonsyndromic Gastric Polyps
Stanford University School of Medicine, Stanford, California
Background
Gastric polyps are rare lesions in the pediatric population. Little is known about their natural history, including their rate of progression to dysplasia and carcinoma. This lack of knowledge hampers efforts to establish uniform treatment and surveillance practices. We present a robust analysis of gastric polyps diagnosed in a single institution and establish their incidence, clinicopathologic spectrum and natural history in pediatric syndromic and nonsyndromic patients.
Methods
We performed a retrospective review of our pathology database to identify patients age 0–21 years diagnosed with gastric polyps from 2000–2015. The histologic slides were reviewed and the diagnoses confirmed by two pathologists. The observed histologic features were correlated with clinical data obtained from the electronic medical record.
Results
Of 4866 pediatric gastric biopsies performed, 64 polyps were identified in 28 patients. Ten polyps were in children age 0–10 years (mean 6.7 years) and 54 were in children age 11–21 years (mean 16.7 years). The polyps consisted of 51 (79.7%) fundic gland polyps (FGPs), 10 (15.6%) hamartomatous polyps, 2 (3.1%) hyperplastic polyps and 1 (1.6%) adenoma. Eighteen patients had a known polyposis syndrome (15 Familial Adenomatous Polyposis (FAP) and 3 Peutz-Jehgers Syndrome (PJS)); 10 patients were nonsyndromic. Low grade dysplasia was present in 14 FAP-associated FGPs and 2 polyps were indeterminate for dysplasia. There was no high grade dysplasia or carcinoma. Of the 50 cases for which the endoscopy report was available, 25 were found in the cardia/fundus, 19 in the body, 4 in the antrum and 2 in the gastroesophageal junction. Polyps were more numerous among older children, ranging from 2 to more than 100 in FAP patients. All polyps measured 0.5 cm or less with the exception of one 1.7 cm hamartomatous polyp in a patient with PJS. Follow-up endoscopies performed in 16 syndromic patients confirm stable polyposis in 12 patients and progressive polyposis in 4 patients. With a mean duration of clinical follow-up in syndromic patients of 86.9 months (range 4.9-185.8), there was no transformation of low grade dysplasia to high grade dysplasia or carcinoma.
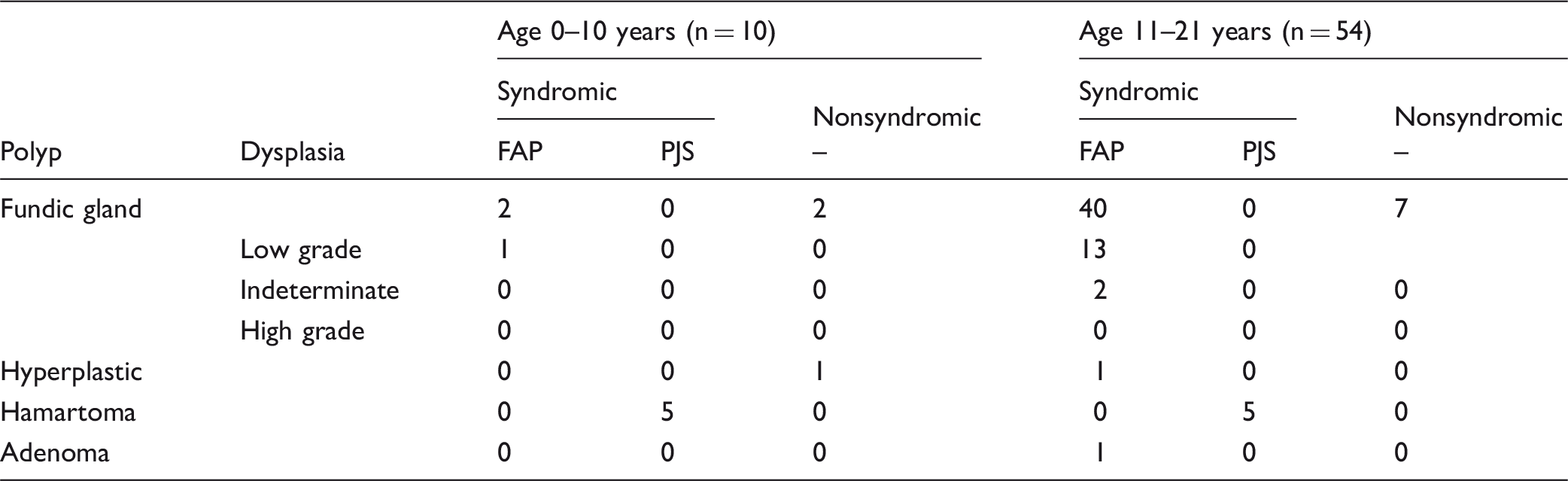
FAP Familial Adenomatous Polyposis; PJS Peutz-Jehgers Syndrome
Conclusion
Gastric polyps are rare in the pediatric population. In our study, most polyps occurred in patients 11–21 years of age. FGPs predominate and low grade dysplasia was exclusively limited to FAP patients. Long-term clinical follow-up demonstrated no progression to high grade dysplasia or carcinoma, suggesting a very low risk of malignant transformation. This analysis adds to the sparse published literature and helps to establish the incidence, clinicopathologic spectrum and natural history of gastric polyps in pediatric syndromic and nonsyndromic patients.
20. Immunophenotypic Study of Ductal Epithelial Differentiation in Pediatric Benign Mesenchymal Hamartomas of Liver
Texas Childrens Hospital, Houston, Texas
Background
Mesenchymal hamartoma is the second most common benign primary hepatic neoplasm in children, typically presenting as a palpable mass with or without elevated alpha fetoprotein levels. Initial lesions are more likely to be solid with multiple malformed bile ducts in a myxoid mesenchymal background. With progressive growth, fluid accumulation within the bile ducts leads to cystic appearance. The epithelium is typically described as biliary type with no variations described in literature so far. We aim to study the pattern of immunophenotypic differentiation in the ductal epithelium of benign mesenchymal hamartomas and its relation to outcome if any.
Methods
This is a retrospective study of all consecutive resections of hepatic benign mesenchymal hamartomas from pathology files of X hospital from 2007–16. One representative block was selected from each tumor and analyzed using immunostains for CK7, CK19, CK20, CDX2, CD56, SOX9, AFP, vimentin and HEPAR; using Leica Bond III autostainer per manufacturer’s instructions. Slides were reviewed in a blind fashion and staining pattern was described for each antibody. Clinical information was obtained from electronic medical records for relevant clinicopathologic correlation.
* Ready to use.
Results
Resected tumors of 7 children (6 M, 1 F) ranging in age from 4 months to 8 years (mean 2.4 years) measured 3.0 to 17.0 cm in greatest dimension. Serum AFP was elevated (700 to 9000 ng/ml) in 3/7 patients; of which one had Beckwith-Wiedemann syndrome. All tumors uniformly showed predominantly biliary type epithelial differentiation in the cystic component as evidenced by strong and diffuse reactivity for CK7 and CK19. Expression of CK20 and CDX2 was seen in 2/7 cases, rare/focal in 1 and diffuse with mucinous differentiation in other. All 7 cases showed strong and continuous nuclear staining for Sox9 in the epithelium lining the cyst wall, similar to the native bile ducts. CD56 staining was variable with strong and diffuse staining in 1 case; and weak, focal staining in 6 cases. Native bile ducts did not show reactivity for CD56. AFP showed patchy weak staining in the entrapped hepatocytic component of the tumor and was negative in the cystic epithelium. Hepar showed focal to patchy staining in the cystic epithelium in 4/7 cases and was negative in the rest. Vimentin was positive in all 7 cases ranging from focal/patchy (4) to diffuse (3). Only 2/7 patients showed abnormal chromosomal analysis; both with CK20/CDX2 positivity. The one with focal staining of CK20 and CDX2 showed characteristic balanced translocation between chromosome loci 2q31 and 19q13; and the one with diffuse staining showed loss of chromosome Y. Remaining 5 patients showed normal chromosomal analysis. All patients with follow up ranging from 4 months to 6 years were alive and free of disease.
Conclusion
Hepatic mesenchymal hamartomas can show divergent epithelial differentiation including both foregut (CK7) and hindgut (CDK20, CDX2) phenotype; and the latter seems to be associated with chromosomal anomalies in the tumor. Divergent epithelial phenotype may provide insights into the pathogenesis of this rare neoplasm but does not seem to affect the outcome and disease free survival in this limited series.
21. Characteristics of Inflammation in Pediatric Steatohepatitis
1Vanderbilt University Medical Center, Dept. of Pathology, Microbiology & Immunology, Nashville, Tennessee
2Vanderbilt University, Department of Biostatistics and Preventive Medicine, Nashville, Tennessee
3Vanderbilt School of Medicine, Nashville, Tennessee
4Vanderbilt Children's Hospital, Dept Pathology, Microbiology & Immunology, Nashville, Tennessee
Background
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are a growing problem in pediatric patients due to the epidemic of obesity. Despite the fact that NAFLD/NASH is increasing, the mechanisms that result in progression of disease and fibrosis are not well understood. Three patterns of steatohepatitis (SH) are recognized in pediatric patients. Type 1 (T1) is similar to that seen in adults with predominantly centrilobulular fat, lobular inflammation, and pericellular fibrosis. Type 2 (T2) shows periportal fat with primarily portal inflammation and periportal fibrosis. The largest group of patients have a mixed pattern with fat throughout the lobule and both portal and lobular inflammation. To our knowledge no data is currently available regarding the composition of the portal and lobular inflammatory infiltrates based on the pattern of SH.
Methods
The pathology database was searched to identify pediatric patients with a liver biopsy with a diagnosis of NAFLD or NASH over a period of one year. Demographic and laboratory data was collected. Children with other medical conditions resulting in liver disease were excluded. Each case was scored using the Brunt classification. All cases had some degree of portal and lobular inflammation, so the greatest weight was given to the zone of fat deposition. Six histologically representative cases of T1, T2, and mixed pattern were selected for immunohistochemistry without knowledge of their body mass index (BMI), age, sex, race, or alanine aminotransferase (ALT), aspartate aminotransferase (AST) or alkaline phosphatase. Each case was dual stained with CD3 and CD68 and two reviewers estimated the proportion of cells to the nearest ten percent. Differences in variables between the three SH patterns were assessed by one-way analyses of variance, t tests, or likelihood ratio chi-squared tests, as appropriate.
Results
Virtually all the cells within the well-stained areas were highlighted by either CD68 or CD3. The inflammatory cells in the portal areas were predominantly CD3 positive for all categories. T2 had the highest percentage of CD3 positive cells (T1, average 47.5%, range 0–80%; T2, average 72%, range 70–100%; mixed, average 67%, range 50–70%). CD68 positive cells were the primary cell type in the lobule (T1, average 70%, range 60–80; T2, average 55%, range 0–80%; mixed, average 68%, range 40–100). The patients with T1 NAFLD had a higher average BMI than patients with T2 histology (41.1 kg/m2 vs. 30.1 kg/m2, p = 0.02) and were older (16.2 years vs. 11.3 years, p = 0.003). All patients had laboratory abnormalities with ALT > AST. Alkaline phosphatase was higher for patients with T2 histology when compared to T1 (103.8 U/L vs. 287.5 U/L, p = 0.001).
Conclusion
Our study suggests that, despite histologic, demographic, and clinical differences, lobular inflammation for T1, T2, and mixed patterns of NAFLD/NASH appears to be CD68 predominant, while portal inflammation appears to be primarily comprised of T-cells. This is interesting given the disparate patterns of fibrosis in T1 and T2. The inflammatory cells involved appear to be different.
22. Rectal Histology in Children Newly Diagnosed with Ulcerative Colitis: The PROTECT Study
1Hartford Hospital, Hartford, Connecticut
2Nationwide Children's Hospital, Columbus, Ohio
3Connecticut Children's Medical Center, Hartford, Connecticut
4Cincinnati Children's Hospital and Medical Center, Cincinnati, Ohio
5Hospital for Sick Children, Toronto, Ontario, Canada
Background
Studies describing pre-therapy histologic changes in children newly diagnosed with ulcerative colitis (UC) are sparse. Prior studies have suggested crypt distortion may be less common than in adults and younger age at diagnosis may have more subtle changes. Our aim was to describe the rectal histology in a well-characterized cohort of children newly diagnosed with UC.
Methods
The PROTECT (Predicting Response to Standardized Pediatric Colitis Therapy) Study enrolled children ≤ 17 years newly diagnosed with UC by standardized clinical, endoscopic and histologic criteria. Central pathology evaluated baseline rectal biopsy for grades of histologic inflammation (I–V), eosinophilic inflammation (1–5) and architectural changes (presence/absence). The relationship between clinical and histologic characteristics was examined.
Results
Rectal biopsies from 368 patients were centrally reviewed. Mean age was 12.9 ± 3.1 years, with 50% female. Extensive/pancolitis at diagnosis was observed in 76%. Fifty-six% of biopsies exhibited > 32 eos/HPF, threshold value for excess eosinophils. Mild acute cryptitis without abscesses (64%) and moderate/marked inflammation with abscesses (25%) were identified. Crypt distortion/atrophy was nearly universal (98%). Architectural changes including surface villiform changes (38%), basal plasmacytosis (53%) and basal lymphoid aggregates (64%) were more variable. Eosinophilic inflammation and grade of acute/chronic inflammation was similar between age (<12 yr, ≥12 yr) and gender. Higher severity of acute inflammation was not associated with increased eosinophilic inflammation. Increased eosinophilic inflammation was associated with presence of basal plasmacytosis (p = 0.01) and lymphoid aggregates (p = 0.00003). More severe grades of acute inflammation were also associated with basal plasmacytosis (p < 0.0001) and lymphoid aggregates (p < 0.0001). Disease distribution, architectural changes, eosinophilic inflammation and grade of acute/chronic inflammation in patients <12 years and ≥ 12 was similar.
Conclusion
Crypt distortion/atrophy, dichotomously scored, was nearly universal in this large cohort of children with newly diagnosed UC. Mild cryptitis was more commonly found than moderate/severe acute inflammation. Increased eosinophilic and higher grades of acute inflammation are associated with signs of chronicity including basal plasmacytosis and lymphoid aggregates at diagnosis. Supported by XXX.
23. Use of Tissue Fixatives on Rectal Biopsies for Subsequent Staining for Acetylcholinesterase in the Diagnosis of Hirschsprung's Disease
University of Minnesota, Department of Pathology, Minneapolis, Minnesota
Background
Central Definitions

Methods
Normal large and small intestine were initially used. After validation, patient samples were used. Eight samples of fresh tissue were fixed in Zeus fixative from 3 to 68 hours and stained for ACHE. Four samples were fixed in formalin, respectively, 0.75, 2.25, 3.5, and 22 hours, and stained for ACHE after sucrose rinse. Six samples were fixed in Zeus fixative followed by formalin fixation and paraffin embedding for calretinin IHC. Prior to frozen section, Zeus fixative was rinsed from the tissue on an orbital shaker, for 30 minutes, in 10% polyvinylporolidone (mw 10,000). Using and orbital shaker, formalin was rinsed from the tissue using 10% sucrose for 15 minutes, followed by 30% sucrose until equilibrated. The ACHE stain was done using a kit produced by Medical and Biological Labs, Co. Ltd. Modifications to the recommended protocol include incubating the substrate 20 minutes, the chromogen 10 minutes, and adding cobalt chloride to the chromogen (final concentration 0.6%).
Results
Both normal gut and HD showed the expected patterns of staining in all samples, 68 hours of Zeus fixation being the longest fixation. Formalin fixation for 3.5 hours or less retained ACHE staining, but activity was lost in the specimen fixed for 22 hours. The calretinin stain performed as expected only in the tissue fixed 1.4 hours in Zeus fixative prior to formalin fixation. The calretinin staining was lost in the other samples (with formalin fixed tissue from the same case serving as a positive control). Polyvinylpyrolidone produced a firm specimen in contrast to Zeus wash and successfully rinsed the fixative from the tissue.
Conclusion
Zeus fixative stabilizes rectal biopsies for an extended period of time allowing biopsies to be held for days prior to ACHE stain. Zeus fixation appears detrimental to the calretinin IHC. Brief fixation in formalin does not affect the ACHE stain, but extended fixation may (more replicates needed). The time limit of Zeus fixation has yet to be determined. Polyvinylpyrolidone is an acceptable substitute for Zeus wash and promotes firmness in the tissue.
24. Pediatric Chondroid Fibrolipoma of the Chest Wall: A Study of Soft Tissue Tumors with Adipose, Fibrous, and Cartilaginous Components
1Children's Hospital of Los Angeles (CHLA), Pathology and Laboratory Medicine, Los Angeles, California
2Children's Hospital of Los Angeles, Department of Surgery, Los Angeles, California
3Children's Hospital of Los Angeles (CHLA),General Pediatric Surgery, Los Angeles, California
Background
Pediatric fibrous and lipomatous lesions of the chest wall are very rare malformative tumors that include mesenchymal hamartoma, chondroid lipoma, collagenous fibroma, lipoblastoma, and fibrous hamartoma of infancy. The presence of a third element, namely cartilage, adds to the diagnostic dilemma of whether the chondroid component is a secondary event such as differentiation or metaplasia versus a primary, neoplastic event. Expression of cyclin D1 (CCND1) has been reported in lipoma, liposarcoma, extraskeletal myxoid chondrosarcoma, chondrosarcoma of bone, and adult chondroid lipoma/chondrolipoma, but not examined in pediatric fibrolipomatous tumors. We hypothesize that mixed cartilaginous, fatty, and fibrous lesions of the chest wall that do not have mesenchymal hamartoma-like features are unique tumors with metaplastic chondroid element. Given its propensity to recur, pediatric chondroid fibrolipoma should be recognized as a distinct entity from adult chondrolipoma.
Methods
A retrospective review of chest wall soft tissue lesions diagnosed at our institution, between 1995 and 2015, was conducted. Hematoxylin and eosin stained sections of the 3 chondroid fibrolipoma cases (1 recurrence in 2 patients) were compared to a series of 1 collagenous fibroma, 4 mesenchymal hamartoma, 8 lipoblastoma, and 4 fibrous hamartoma of infancy. Immunohistochemical staining was performed in each chest wall neoplasm with anti-cyclin D1, which is expressed in pediatric mesenchymal hamartoma but not chondroid fibrolipoma, contrary to adult chondrolipoma. Additional immunohistochemical staining with S100, β-catenin, CD99, and CD34 serves as comparative tests.
Results
The 20 cases consisted of 9 females and 9 males with 1 recurrent and 1 excised cases [mean age 4.2 years, range 3 weeks to 15 years]. All mesenchymal hamartoma cases showed strong positivity for cyclin D1 expression. All chondroid fibrolipoma, collagenous fibroma, mesenchymal hamartoma, lipoblastoma, and fibrous hamartoma of infancy demonstrated no cyclin D1 expression. The chondroid fibrolipomatous tumor arising in a three year old female consisted of immature cartilage, fat, and fibrous tissue. It recurred twelve years later and showed features of lipofibromatosis, without a cartilaginous component. The other case arose in a seven year old female and consisted of immature cartilage, fat, and fibrous tissue.
Conclusion
Pediatric chondroid fibrolipoma of the chest wall appears to be a histologically unique lesion that may be more akin to lipofibromatosis and tends to recur. It is distinct from other pediatric chest wall tumors as well as adult chondroid lipoma/chondrolipoma, which does not recur.
25. A Report of Two Infantile Sarcomas with NTRK1-TPM3 Rearrangements
Seattle Children's Hospital, Seattle, Washington
Background
Infantile fibrosarcoma is a locally aggressive tumor of young children, occurring almost exclusively under the age of 2 years. It is characterized cytogenetically by non-random gains of chromosome 8, 11, 17 and 20, and more specifically by an ETV6-NTRK3 gene fusion. NTRK3 is one member of the family of tropomyosin-receptor kinases, along with NTRK1 and NTRK2. A small molecular inhibitor to the broad TRK family is available and in clinical trials for patients with unresectable and/or recurrent TRK associated tumors. Gene fusions involving the other TRK family members have been implicated in a variety of neoplasms, including colorectal carcinoma and papillary thyroid carcinoma, and more recently NTRK1 mutations have been described in fibrous hamartoma of infancy and juvenile lipofibromatosis.
Methods
We describe two cases of ETV6 negative soft tissue sarcomas occurring in infants under the age of 5 months, for which molecular characterization was performed using a targeted next-generation sequencing (NGS) platform developed for the identification of therapeutic mutations in cancer.
Results
Patient 1 presented at 3 months of age with a small lump on his arm. Resection at 5 months of age showed a hypercellular neoplasm characterized by primitive, ovoid cells in a myxoid stroma with focally prominent nuclear palisading. Mitoses numbered 3 per 10 high power fields. No necrosis was seen. Immunohistochemical studies were negative for CD34, S100, SMA, myogenin and EMA. Florescence in situ hybridization (FISH) was negative for ETV6 rearrangement, but trisomy 8, 17 and 20 were identified by conventional karyotype.
Patient 2 presented at birth with a large right lower leg calf mass. Resection at 3 weeks of age demonstrated a hypercellular spindle cell lesion, characterized by elongated spindle cells arranged in fascicles. Mitoses numbered 2 per 10 high power fields. No necrosis was present. Immunohistochemical studies were negative for CD34, S100, SMA, desmin, myogenin, EMA and CD99. Cytogenetic studies lacked evidence of an ETV6 rearrangement but identified trisomy 8 by FISH. A second resection revealed increased mitotic activity (7 per 10 high power fields) and changing morphologic patterns, with increasing myxoid stroma and primitive, ovoid cells similar to those seen in Patient 1. Both patients developed multiply recurrent disease, and tumor tissue was submitted for molecular analysis by NGS to identify potential therapeutic mutations. An NTRK1-TPM3 rearrangement was identified in each tumor.
Conclusion
We report two locally aggressive soft tissue tumors in infants which mimicked congenital infantile fibrosarcoma, but lacked evidence of the ETV6 gene rearrangement characteristic of infantile fibrosarcoma. Instead, these two tumors with somewhat disparate morphologic appearances showed a novel NTRK1-TPM3 gene fusion amenable to targeted therapy. Recognition of the growing role of the TRK family of genes in pediatric soft tissue neoplasia is important to both diagnosis and treatment for young children with these potentially aggressive tumors.
26. TLE1 is Expressed in Congenital Infantile Fibrosarcoma/Cellular Congenital Mesoblastic Nephroma
Children's Hospital Los Angeles, Los Angeles, California
Background
TLE1 is a highly sensitive but relatively non-specific marker for synovial sarcoma (SS), with positivity described in a subset of other sarcomas. Its expression has not been well studied in congenital infantile fibrosarcoma (CIFS), which may histologically mimic SS and overlap rarely in clinical presentation. CIFS is a rare soft tissue tumor of infancy characterized by an EVT6-NTKR3 gene fusion akin to its renal counterpart cellular congenital mesoblastic nephroma (CMN). This study attempts to characterize TLE1 expression in both entities through immunohistochemistry (IHC) analysis.
Methods
We reviewed 8 archived CIFS and 7 cellular CMN cases. TLE1 IHC was performed on formalin-fixed paraffin-embedded tissue blocks for all cases. Four classical CMNs were included for comparison.
Results
The 15 cases (median age = 10 weeks, range = 2 days–4 years) consisted of 5 males. Evidence of ETV6 rearrangement was available via prior FISH, RT-PCR, or karyotype studies in 8 of the 15 cases. Of the CIFS subset, 5 contained characteristic cytogenetic abnormalities, 2 had unknown cytogenetic status, and 1 was negative by FISH. Within the cellular CMN subset, 3 were confirmed by cytogenetic studies and 4 were untested. Thirteen of 15 cases expressed TLE1 with the following results: positive in all 5 CIFSs with typical cytogenetic abnormalities, with diffuse moderate to strong intensity; positive in 1 of 2 CIFSs with unknown cytogenetic status; and negative in the FISH-negative CIFS. All 7 cellular CMNs showed diffuse moderate to strong staining for TLE1. Histologically, CIFS and cellular CMN generally showed dense sheets and whorls of short plump uniform spindle cells. The 2 CIFS cases that lacked TLE1 staining contained more well-differentiated fibroblasts and myofibroblasts arranged in long sweeping fascicles. Incidentally, 4 classical CMNs (median age = 4 weeks, range = 1 day–7 weeks) showed negative to patchy weak TLE1 staining.
Conclusion
TLE1 is often expressed in CIFS and cellular CMN with a diffuse and strong staining pattern, suggesting that it cannot be relied upon to distinguish SS from CIFS. Evaluation for both ETV6 and SYT gene arrangements may be necessary to discriminate these two soft tissue tumors, especially when occurring in infants and younger children. Two TLE1-negative CIFS cases lacked confirmatory ETV6 rearrangements and showed less characteristic histology, and may represent different entities. By comparison, TLE1 was negative or weakly expressed in classical CMN. Thus subjective interpretation may limit its utility as a distinguishing marker from cellular CMN. In summary, this study expands the spectrum of TLE1-positive tumors to include CIFS/cellular CMN.
27. Beta-Catenin Immunohistochemistry and Wnt Pathway Activation in Medulloblastoma: An Imperfect Correlation
Boston Children's Hospital, Boston, Massachusetts
Background
Molecular substratification of medulloblastomas is recommended by the 2016 edition of the WHO Classification of the Tumours of the Central Nervous System. The methods used for their classification are immunostains that are surrogate for pathway activation (beta-catenin, GAB1, YAP1), fluorescence in situ hybridization (Myc, MycN, isochromosome 17), and institution-dependent molecular platforms able to identify numerous genetic alternations. Recognizing Wnt medulloblastomas is important, as their survival is higher than 90%, and the patients may benefit from less aggressive therapy. This study presents a correlation of beta-catenin immunostain results with the array comparative genomic hybridization (CGH) and targeted exome sequencing of 300 genes (Oncopanel) data.
Methods
Thirty-two consecutive medulloblastomas from 2012–2016 were retrieved from the archives of our institution. The hematoxylin-eosin-stained slides, immunohistochemical stains (synaptophysin, glial fibrillary protein, INI1 and beta-catenin), array CGH and Oncopanel data were reviewed. Any discordance between the beta-catenin immunostain and Wnt pathway activation detected by molecular techniques was discussed at the molecular brain tumor board.
Results
Of 32 medulloblastomas, 20 had classic, 3 had desmoplastic nodular, and 11 had anaplastic/large cell morphology. There were 3 Wnt-, 5 SHH-activated, 8 class 3, and 16 class 3 or 4 medulloblastomas. The beta-catenin immunostain was initially reported as negative in all cases. However, the presence of monosomy 6 and CTNNB1 c.94G > T(p.D32Y), exon 2 mutations in 3 tumors, consistent with Wnt pathway activation, prompted the re-evaluation of beta-catenin immunostain. In these 3 tumors, 1–2% of the nuclei expressed beta-catenin. Two class 3 medulloblastomas also showed rare beta-catenin-positive nuclei.
Conclusion
Wnt pathway activation in medulloblastomas does not always correlate with diffuse nuclear expression of beta-catenin. Nuclear expression in very rare cells (as opposed to diffuse) should prompt an investigation for presence of monosomy 6 and/or beta-catenin mutations, in order to confirm or rule out Wnt pathway activation.
28. Decreased Expression of Soluble fms-like Tyrosine Kinase 1 (sFlt1) in Abnormal Placentation
Beth Israel Deaconess Medical Center, Boston, Massachusetts
Background
Abnormal placentation (placenta accreta, increta, and percreta) is the leading cause of life-threatening post-partum hemorrhage. Antenatal diagnosis is typically accomplished by sonographic evaluation; however, imaging is limited as a screening methodology, and definitive diagnosis depends on histologic examination showing placental invasion into the myometrium. In these cases, trophoblasts can be seen invading myometrial blood vessels, suggesting unbalanced angiogenic activity and possible increased perfusion from these larger vessels. Soluble fms-like tyrosine kinase 1 (sFlt1) is an anti-angiogenic protein that has been shown to be up-regulated in preeclampsia, a state associated with impaired placental angiogenesis, shallow implantation, and hypoperfusion. The primary objective of this study was to characterize expression of sFlt1 in invasive placentation. We hypothesized that areas of placental invasion would have low expression of sFlt1 compared to areas of normal placentation.
Methods
This is a retrospective study of peripartum hysterectomy specimens which received a pathologic diagnosis of abnormal placentation at a single institution from January 1995 through June 2015 (n = 38). Histologic review of existing (H&E) slides was undertaken to choose appropriate cases. Cases with multigestation, fragmented/partially delivered placentas, or clinical history of preeclampsia were excluded. Twelve cases with ideal histology were selected. Formalin-fixed, paraffin-embedded tissue sections were stained for sFlt1, as well as cytokeratin and Masson Trichrome for definitive diagnosis and characterization of placental invasion. Among the twelve cases, fifteen foci of deep invasion and sixteen foci of non-invasion (accreta-like) were identified. Within each focus, the five high-power fields with highest sFlt1 staining within the chorionic villous trophoblasts were scored from 0 to 3, 0 corresponding to no staining and 3 being the most intense staining, using intermediate implantation site trophoblasts as an internal control (placental bed trophoblasts show high expression of sFlt1, which likely serves as a barrier function against vascular leak or excess angiogenesis in normal placentation). Five control cases were originally selected, however one case showed markedly increased sFlt1 staining compared to the others. Further investigation into the clinical history showed the patient had been diagnosed with pre-eclampsia. This case was subsequently eliminated from analysis. The sum of scores from the five high-power fields in foci of deep invasion, non-invasion (accreta-like), and control cases were compared. Each focus was considered a separate data point, assuming that expression in areas separated by at least 2–3 mm were independent.
Results
Chorionic villous trophoblasts displayed significantly decreased expression of sFlt1 in areas with histologic placental invasion compared to areas of normal placentation (ANOVA, p < 0.003).
Conclusion
Expression of sFlt1 is diminished in abnormal placentation, suggesting a pro-angiogenic state in areas on myometrial invasion. Further study is warranted to elucidate the mechanistic role of sFlt1 and its potential utility as a marker of abnormal placentation.
29. CISH Localization of Placental miR 518b Expression in Amniotic Fluid Infection
1British Columbia Children's Hospital, Vancouver, British Columbia, Canada
2UBC Medical Genetics and British Columbia Children's Hospital Research Institute, Vancouver, British Columbia, Canada
Background
Placental microRNAs (miRs) are detectable in maternal serum and expression patterns may change in some pathological states, implying utility as a diagnostic marker. A miR-based maternal serum test for amniotic fluid infection (AFI) could be clinically useful. Our qRT-PCR studies on formalin-fixed, paraffin-embedded (FFPE) placental tissue show miR 518b is dysregulated in AFI. Since placental miRs in maternal serum are primarily derived from trophoblast, localization of 518b expression would inform its potential applicability as a serum marker of AFI. Here we report the results of a pilot study assessing 518b expression in AFI placenta using chromogenic in situ hybridization (CISH).
Methods
This study is approved by the research ethics board. Sections of archival placental disc are taken from 5 normal term (NT; 37–41 weeks gestational age, average 38.8), 4 AFI term (AFIT; 38–41, 39.6; average maternal inflammatory response stage 2.4/grade 2, fetal inflammatory response stage 2/grade 1.6), 5 non-inflamed (clinical acute abruption) preterm (NP; 25–28, 26.6), and 5 AFI preterm (AFIP; 26–27, 26.4; MIR 2.8/2, FIR 1.8/1.2) cases. The CISH protocol is based on previously described methods and employs digoxigenin-labelled anti-miR 518b probe with DAB chromogen visualization. U6 snRNA probe is used for protocol optimization and positive control. Negative controls are scrambled miR probe and exclusion of probe. Staining intensity is semi-quantitatively graded as absent, weak, moderate, or strong (strong = intensity of optimized U6 NT staining) and distribution as rare (<10% of cells), focal (10–50%), or diffuse (>50%).
Results
Syncytiotrophoblast shows mostly absent 518b staining in all 4 groups, although weak/moderate focal staining is seen adjacent to the maternal surface. Increased intensity in all other cellular compartments is also seen near the maternal surface suggesting fixation effect. Amnion shows moderate diffuse staining in the AFIT/AFIP groups compared to weak rare staining in NT/NP. Extravillus trophoblast is moderate diffuse in AFIT/AFIP while weak diffuse in NT/NP. Villus stromal staining is weak diffuse in all groups but a subset of AFIT/AFIP shows focal moderate intensity while NT/NP is consistently weak. All groups show weak diffuse cytotrophoblast and weak/moderate focal fetal endothelium staining. U6 shows increased staining in AFIT/AFIP compared to NT/NP. Scrambled probe shows non-specific focal cytoplasmic staining. Omission of probe shows no staining.
Conclusion
CISH-based expression analysis demonstrates the majority of miR 518b expression occurs in cellular compartments other than trophoblast implying 518b is a suboptimal candidate as a maternal serum marker for AFI. Increased staining observed towards the maternal surface suggests fixation rate plays a role in miR 518b preservation and thus overall expression levels may be underestimated in archival tissue. AFI-associated increased 518b expression in amnion, extravillus trophoblast, and villus stroma may account for the qRT-PCR-based study results.
30. Basal Chronic Villitis is Associated with Hypertensive Disorders of Pregnancy
University of Rochester Medical Center, Rochester, New York
Background
Hypertensive disorders (HD) of pregnancy are a common cause for preterm delivery. Chronic villitis of unknown etiology (CVUE) is associated with IUGR, pre-eclampsia and IUFD. The authors hypothesize that chronic inflammatory lesions of the placenta, especially basal chronic villitis (CV), are associated with HD.
Methods
In this retrospective study, the LIS was queried over a 12 year period to identify placentas processed with or without the clinical history of HD using the keywords “ECLAMPSIA”, “HYPERTENSION”, “HTN”, or “HELLP” and with the final diagnosis using key phrase “CHRONIC VILLITIS”. Pathology reports of placentas with and without HD were reviewed and divided into those with multifocal CV and basal CV. Comparisons between subgroups with and without HD were made using the chi square statistic with significance set at P < 0.05 (www.socscistatistics.com).
Results
20,862 third trimester placentas were identified. 15.5% of these placentas had some form of CV and 16.8% were associated with HD. CV was present in 18.0% and 15.2% of placentas associated with or without HD, respectively, a significant difference (P < 0.0001). Similarly, basal CV was present in 6.8% and 4.6% of placentas associated with or without HD, respectively, also a significant difference (P = 0.000002). However, in placentas with multifocal CV there was no significant difference between cases and controls (7.6% with HD vs. 6.7% without HD, P = 0.09).
Conclusion
Basal CV is more strongly associated with HD in pregnancy than multifocal CV. This finding may be due to an abnormal basal plate in HD with reaction to vascular lesions such as maternal and hypertensive decidual vasculopathies.
31. Placental Chorionic Surface Arterial Networks: Development of the Surface Distribution Networks from 24 Weeks through Term
1Department of Obstetrics/Gynecology, New York Methodist Hospital, Brooklyn, New York
2St. George's University School of Medicine, Brooklyn, New York
3Placental Analytics, LLC, New Rochelle, New York
4New York Methodist Hospital, Brooklyn, New York
Background
We have previously demonstrated a method for the reliable reproduction of the placental chorionic surface arterial networks (PCSAN), and that such networks differ among term singleton births with and without diabetes, and in monochorionic twins with and without birth weight discordance. Here we present a preliminary analysis of preterm and term placentas from a unique birth cohort in which all placentas are referred for pathologic examination.
Methods
According to a previously published protocols, 46 digital photographs of placental chorionic surfaces were obtained and the PCSAN were traced by a single observer (AS). The traced images were analyzed by a dedicated algorithm that calculated a range of PCSAN features including branch point numbers, network lengths, branch angle and vessel tortuosity. Non-parametric test (Spearman's correlation, Mann-Whitney U test) were used to compare distributions of these novel variables (means and standard deviations, SD) as a function of gestational age, with p < 0.05 signifcant.
Results
Branch angle (mean and SD) were strongly correlated with GA (r = 0.48, p = 0.001; r = 0.35, p = 0.02, respectively). Mean tortuosity (r = 0.33, p = 0.02) and the distance from arterial end points to the chorionic disk edge (mean, r = 0.37, p = 0.01; SD r = 0.33, p = 0.03) were correlated with GA. Branch point numbers and the distances between arteries and veins were constant across GA. The correlation of PCSAN features differed by gender, as the 2 branch angle measures were only correlated with GA in males (r = 0.59, p = 0.003; r = 0.64, p = 0.001, respectively), and mean tortuosity, and mean and SD of distance from arterial endpoints to the perimeter were only correlated with GA in females (r = 0.48, p = 0.02; and r = 0.43, p = 0.04, r = 0.44, p = 0.04, respectively).
Conclusion
Specific features of PCSAN differ by GA, and these differences are gender dimorphic. These may provide yet another means by which the fetal enviroment differs by gender and account for the gender dimorphism of fetal programming.
32. Placental Expression of the Forelimb Patterning Transcription Factor MEIS2 in Trisomy 15
British Columbia Children's Hospital, Vancouver, British Columbia, Canada
Background
Little is known about the phenotype of trisomy 15 during embryogenesis but observations of first trimester human trisomy 15 embryos have identified meromelic changes in the upper limbs. These changes are similar to those observed in animal studies investigating the effects of overexpression of Meis2, a signaling transcription factor expressed transiently during proximo-distal forelimb development. The high degree of conservation and almost identical role of Meis homologues in different organisms implies the human homologue functions in the same manner. Human MEIS2 is located on chromosome 15 and increased gene dosage resulting from trisomy 15 may lead to protein-level MEIS2 overexpression and meromelia. Directly assessing MEIS2 expression in human embryonic arm would be exceedingly difficult; however, MEIS2 has been reported as consistently expressed in first trimester placental villus stroma. This study addresses whether gene dosage effect might underlie meromelia in trisomy 15 by comparing MEIS2 expression in placenta from trisomy 15 and euploid spontaneous abortions.
Methods
Research ethics board approval is obtained prior to study initiation. 30 serial first trimester spontaneous abortions with isolated trisomy 15 (T15) and 30 serial first trimester euploid controls (EC) are retrieved from hospital archives. IHC is automated with anti-human MEIS2 antibody (Sigma clone 1H4, 1:350 dilution). Spleen is the positive control and mature skeletal muscle the negative control. MEIS2 IHC staining is graded on a semiquantitative scale of 1 = weak (spleen), 2 = moderate, and 3 = strong. Non-nuclear staining is ignored. 100 positive villus stromal cells in viable non-stem villi are assessed and the scores averaged per section. The average section scores for T15 and EC are compared using Student’s t-test.
Results
MEIS2 nuclear staining is consistently seen throughout all assessed villi but shows variable intensity between nuclei within villi of both T15 and EC tissue. The average T15 MEIS2 IHC score is 2.15 (95% confidence interval 2.00–2.30) and the average EC score is 1.93 (95% confidence interval 1.77-2.09). The difference between the average scores for T15 and EC is marginally statistically significant (p = 0.0485). MEIS2 expression is also present in decidual stroma (strong), extravillus trophoblast (moderate), fetal endothelium (weak), and cytotrophoblast (weak). Syncytiotrophoblast and Hofbauer cells appear negative. Spleen exhibited positive MEIS2 nuclear staining (grade 1) and skeletal muscle nuclei were negative in keeping with previous reports.
Conclusion
Average MEIS2 expression is increased in T15 first trimester placental tissue compared to EC but the difference is marginal. Unadulterated gene dosage effect is expected to increase MEIS2 expression by 50%, which in this study would give an average T15 score of 2.90, but this is not seen. This result indicates that the extra copy of MEIS2, at least in placental villus stromal tissue, does not lead to a proportional increase in MEIS2. Extrapolation of this result to the developing embryonic arm suggests an alternative mechanism underlies meromelia in trisomy 15.
33. Placental Histological Study in Post-Partum Preeclampsia: Is the Placenta Essential to Preeclampsia?
UT McGovern Medical School at Houston, Houston, Texas
Background
Preeclampsia (Pre-E) is a syndrome specific to gestation that affects 2–7% of pregnancies. It is thought to be related to ischemic alterations of the placenta. The only known cure is delivery. Post-partum Pre-E is the occurrence of the syndrome 48 h to 6 weeks after delivery. It is not explained by our current understanding of the physiopathology. Histopathological studies are valuable tools in understanding placental disease. The objective was to perform a histological study of the placenta of women who will later present with postpartum preeclampsia and compare them to the placentae of women with preterm and term preeclampsia as well as normotensive subjects (NT).
Methods
This is a case-control study, comparing the placental histologic study in 4 groups of 30 patients (post-partum Pre-E vs early antepartum Pre-E, late antepartum Pre-E, NT subjects). Patient with Pre-E were identified on an established registry of hypertensive disorders of pregnancy. NT control subjects were identified by through ICD codification. Placental examinations were performed by one board-certified pathologist, to avoid observer bias. Data are expressed in mean ± standard deviation or percentage (absolute) and analyzed by the ANOVA or Chi2 test as appropriate.
Results
There was no difference in age, ethnic group, BMI, pre-gestational diabetes, primiparity and IVF between groups. Patient with post-partum Pre-E had more gestational hypertension (13.3% (4) vs none in controls, p = 0.04), mean gestational age at delivery was 36.3 ± 3.6 weeks (early Pre-E: 30.4 ± 3.0, p < 0.01; late Pre-E: 36.9 ± 1.6, p = 0.9; NT: 38.7 ± 1.9, p < 0.01) and mean birthweight was 2654.0 ± 798.7 grams (early Pre-E: 1468.6 ± 602.5, p < 0.01; late Pre-E: 3133.5 ± 714.2, p = 0.06; NT: 3112.0 ± 579.1, p = 0.07). Placental weight of post-partum Pre-E was 479.0 ± 152.7 grams (early Pre-E: 277.0 ± 125.8, p < 0.01; late Pre-E: 521.3 ± 144.1, p = 0.7; NT: 502.4 ± 97.7, p = 0.9). Forty percent had acute deciduitis (early Pre-E: 5.7 (2), p < 0.01; late Pre-E: 16.7 (5), p = 0.046; NT: 3.2 (1), p < 0.01), 39,4% (13) had abnormal maturation of the villi (early Pre-E: 77.2 (27), p < 0.01; late Pre-E: 26.7 (8), p = 0.3; NT: 3.2 (1), p < 0.01), 18.2% (6) had decidual arteriolopathy (early Pre-E: 34.3 (12), p = 0.13; late Pre-E: 10 (3), p = 0.35; NT: 9.7 (3), p = 0.33) and 9.1% (3) had villous ischemia and infarction (early Pre-E: 51.4 (18), p < 0.01; late Pre-E: 13.3 (4), p = 0.6; NT: 16.1 (5), p = 0.4). There was no difference in chronic deciduitis, decidual necrosis, retroplacental hemorrhage, meconium staining, chorangiosis and acute inflammation of the chorionic plate between groups.
Conclusion
Gestational hypertension (GH) is more frequent in women with postpartum Pre-E. Preeclampsia has a wide spectrum of presentation and GH might be the early stage of the disease, that will later be clinically expressed in the postpartum. As expected, more severe vascular and ischemic placental lesions were found in patients with early onset Pre-E. However, there were no significant differences for decidual arteriolopathy and villi ischemic changes between postpartum Pre-E, late onset Pre-E and the controls. This suggests that late-onset and postpartum Pre-E are maternal disease and share a common physiopathology were the placenta plays little or no role.
34. Cystic Crypt Changes in Midtrimester Fetal Vermiform Appendix: An Unrecognized Transient Histologic Feature
1The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania
2Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania
Background
Lewis, and subsequently Huff, have briefly described a transient “microcystic cryptitis” observed microscopically in some of the vermiform appendices (VA) of early to late midtrimester fetuses at autopsy. To this date, this phenomenon remains unexplored, and whether it plays a role in the physiologic developmental process of the gut, or represents a response to a certain stimulus is still unclear. In this study, we aimed to describe these crypt changes (CC), their pattern of occurrence in relation to gestational age, and determine if they are related to infection.
Methods
All available H&E-stained sections of VA from perinatal autopsies performed from year 2000 to 2015 were reviewed. The presence or absence of CC and the different histomorphology of these crypts were documented. In addition, the autopsy reports were reviewed for evidence of amniotic fluid or fetal systemic infection including acute chorioamnionitis, chronic villitis with plasma cells, and fetal sepsis (as documented by blood culture and/or organ histopathologic findings). The data were analyzed using descriptive statistics, and logistic regression analysis was employed to determine the relationship between the CC and incidence of infection.
Results
A total of 345 VA from 15 to 40 weeks gestational age fetuses, of which 161 (46.7%) were males and 184 (54.3%) were females, were included; greater than half of the cases (n = 184; 53%) were from 19–23 weeks gestation. CC were found in 202 (58.5%) of VA, and were characterized by one or a combination of two or more of the following features: crypt dilatation with or without irregularity of the lumen, crypt dilatation demonstrating semi-attenuated epithelium with intraluminal apoptotic debris and some inflammatory cells especially intact and degranulated eosinophils, and foci of swirled spindled and polygonal mesenchymal cells, which may rarely be associated with calcifications or multinucleated giant cells. Occasionally, VA also showed nodular aggregates of lymphocytes adjacent to the distorted crypts. The CC begin to appear at 17 weeks gestation, peak at 20–25 weeks gestation (66 to 82% of VA showed CC during this time), with a steady decline beyond 28 weeks gestation. Among the VA with CC, 95 (47%) had coexisting infection and 107 (53%) had none. Among the VA without CC, 65 (46%) had coexisting infection and 78 (54%) had none. Based on Nagelkerke pseudo R-square, only 0.4% of cases with CC may be attributable to infection, and the presence of CC is not related to the incidence of infection.
Conclusion
Our study described the underrecognized CC of the developing fetal VA, which may be used as a histologic marker of mid trimester VA because of its distinct temporal pattern of occurrence. The CC did not seem to be affected by the presence or absence of amniotic fluid or fetal systemic infection, which favored their being a part of the normal gut development process, and perhaps a potential role in the assembly of mucosa-associated lymphoid tissue. Nonetheless, recognition of the normal histomorphologic spectrum of these crypts is important because of the similarity of these CC to “true cryptitis” found in the setting of an infectious or inflammatory process, which may lead to erroneous diagnosis, particularly when evaluating VA of extremely preterm neonates.
35. Human Cytomegalovirus Infection Detected by Immunohistochemical Staining is an Uncommon Finding in Fetal Growth Restriction
OHSU, Portland, Oregon
Background
Cytomegalovirus (CMV) is a common cause of congenital infection worldwide (1.0%). Maternal primary infections are more likely to lead to congenital transmission and birth defects. Their placentas may have chronic villitis and PCR studies suggest 10% of chronic villitis cases may be positive for CMV. A recent study suggested placental CMV infection may also be a
Methods
Retrospective immunohistochemical study of formalin-fixed paraffin-embedded placental and umbilical cord tissue sections from seven cases of positive primary maternal CMV infection (2 had FGR), 10 cases of FGR without maternal CMV serology results, 48 cases of chronic villitis, and 48 cases without chronic villitis. Anti-CMV monoclonal IgG clones (CCH2, DDG9, Dako) routinely used by our laboratory to diagnose positive CMV infections in various tissue biopsies were employed to test this placental cohort.
Results
Two of seven primary maternal CMV infection cases showed positive CMV immunostaining in placental sections (cytotrophoblasts) (30%), consistent with the expected frequency of congenital transmission. Two of the maternal CMV cases were diagnosed with FGR, one of these two cases immunostained for CMV; the remaining 10 cases of FGR were all negative for CMV (1/12 positive, 8%). None of the remaining 96 cases were positive for CMV by immunohistochemistry, including 0/48 cases of chronic villitis. PCR testing for CMV has not yet been performed.
Conclusion
The anti-CMV clones employed by our clinical laboratory (and likely many others around the world) have been tittered to achieve both high sensitivity and specificity. We conclude the high frequency of CMV immunostaining in the recently published FGR study most likely represented false positive staining. Moreover, our data suggest CMV immunoreactivity is also a rare finding in chronic villitis (<2%).
36. Quality Improvement for Pediatric Surgical Pathology- 7 Years of Data with Proposed Benchmarks and Recommendations to Reduce Pathologist Error
University of Utah School of Medicine, Salt Lake City, Utah
Background
We are unaware of benchmarks for diagnostic performance in pediatric surgical pathology. We therefore reviewed error rates over a seven-year period for seven pathologists at different experience levels.
Methods
Over a seven-year period, we reviewed 1,108 of 36,281 surgical pathology cases that were amended or tagged by the primary or review pathologist with a quality code indicating a major quality or diagnostic problem. A previously described coding system for surgical pathology report facilitated the review process.
Results
Of the 1,108 cases reviewed, 69 were due to pathologist error. The most common categories of pathologist error with potential clinical significance were: frozen section diagnosis of CNS lesions (8, of which 7 were low grade tumors or benign lesions), sign out of slides from wrong case (7), missed micro-metastases in lymph nodes (3), missed pathogens (3), and frozen section diagnosis of Hirschprung disease (3). The average error rates for 2 pathologists with 0– 5years' experience were 4.1 and 3.9 errors/1,000 cases. The average error rates for 2 pathologists with 5–10 years' experience were 0.2 and 1.75 errors/1,000 cases. The average error rates for 5 pathologists with over 10 years' experience were 0.64, 0.70, 2.5, 3.1, and 3.8 errors per 1,000 cases.
Conclusion
Procedural changes to address common causes of error included second pathologist review for (1) negative lymph nodes affecting stage of malignant neoplasm, (2) frozen sections on CNS lesions and (3) frozen sections for Hirschprung’s disease. Bar coding of slide labels and mandatory scanning prior to dictation is anticipated to decrease errors due to transposing slides. Our data suggest that pathologist errors decrease with experience. In our practice, 4 errors or more per 1,000 cases triggers a review and possible implementation of a focused process change.
37. Inpatient Genetic Test Review Benefits from Lab Determined Criteria
1Vanderbilt Children's Hospital, Nashville, Tennessee
2Vanderbilt University Medical Center, Nashville, Tennessee
3Vanderbilt University, Nashville, Tennessee
Background
Genetic test review (GTR) results in better test utilization and cost savings. Our institution implemented a reference test review on all germline genetic testing in both the inpatient (IPT) and outpatient (OPT) setting as part of a quality improvement process. GTR in the IPT population is important because of capped reimbursement. Developing criteria for IPT testing could be a helpful utilization management strategy. With high numbers of genetic tests ordered on pediatric patients, GTR is of special interest to laboratories serving this population.
Methods
Our laboratory services a pediatric and an adult tertiary care hospital. Genetic tests ordered on both IPTs and OPTs come through our laboratory and are identified for GTR by their purple top tube (EDTA) and ordered test name. Once captured for GTR, order information is entered into a database. All tests are reviewed by a genetic counselor (GC) and all test changes, cancellations, and IPT orders are also reviewed by a pathologist. The team records data on clinical indication for testing, appropriateness of the test, vendor lab, cost, and patient demographics. In addition the GC selects one or more of the following criteria for IPT testing: 1) Genetic testing will result in a change in patient care once resulted (CRI1); 2) Genetic testing will be resulted during the patient stay(CRI2); 3) The patient is likely to die during the hospital stay and testing is necessary to confirm or rule out a diagnosis for the family and future family planning (CRI3); 4) Genetic testing will likely uncover/confirm the diagnosis which is requiring hospitalization (CRI4); 5) Other (CRI5).
Results
During a 5 m review period, 698 orders were reviewed and 69 reviews were ordered on IPTs. Of IPT reviews, 14 were out-of-scope of the GTR team (4 cancer based, 6 pharmacogenetics, and 3 other). In total 55 test orders were within the scope of the project. Two reviews were on adult patients and 53 were on pediatric patients (age < 18 yrs). Nine patients had more than one order. The age range of patients was 1 day to 26 years. Thirty-four orders had no change, 9 reviews resulted in a test change, 7 tests were cancelled and 5 tests had a vendor change. Cancellation occurred for different reasons, including a duplicate test was ordered, results of a pending test could guide further work-up, or the patient was stable and work-up was appropriate for the OPT setting. The genetic counselors selected CRI1 in 31 cases, CRI2 in 18, CRI3 in 8, CRI4 in 26, and CRI5 in 2 cases. Pathology staff approved all testing that was sent. The total saved by the review process was $13,500-17,500. (Results are reported as a range to account for reflexed testing.) Thirty-eight percent (21 of 55) of the IPT reviews resulted in alterations of the original orders, versus 21% (137 of 538) of OPT reviews during a similar time frame. Cancellations were 2.5 times as frequent in the IPT group versus the OTPT group, 13% (9 of 55) versus 5% (26 of 538).
Conclusion
Pediatric patients make up the majority of IPTs on whom genetic testing is ordered, and therefore, this group is of particular interest when planning utilization management efforts. Lab selected criteria can help identify testing which is not only indicated for the patient, but appropriate for the INPT setting.
38. Diagnostic Yield and Cost Analysis for Sequential Sequencing and Deletion/Duplication Genetic Testing in a Pediatric Referral Center
1University of Washington/Seattle Children's Hospital, Seattle, Washington
2Seattle Children's Hospital, Seattle, Washington
Background
As test methods for inherited conditions become increasingly complex, pediatric hospital laboratories must play a greater role in genetic test selection and utilization management (UM). Consequently, pathologists and laboratory genetic counselors can now centralize information amongst varied medical specialties regarding testing outcomes and costs for their patient population. This coordination is particularly valuable for genetic conditions in which a significant proportion of pathogenic variants would not be detected by sequencing methods alone, and may require a second, separately ordered method. Such is the case for larger deletions and duplications (del/dups), currently best characterized by methods such as multiplex ligation-dependent probe amplification (MLPA) or array comparative genomic hybridization (aCGH), excluding select newer next generation sequencing (NGS) platforms.
Methods
We assessed the frequency of positive (pathogenic) results and cost savings for UM team-guided sequential testing in inherited conditions for which either point mutations or deletions alone could be diagnostic (i.e. autosomal dominant or x-linked disorders). Second, for common single gene-associated conditions tested by separate sequencing and del/dup methods (reflexive or concurrent), we compared our diagnostic findings profile to literature estimates. Third, for rarer (infrequently ordered) genes in which the proportion of probands with pathogenic del/dups relative to sequence variants is less well known, we assessed the diagnostic yield of sequential testing as described above.
Results
Query of our UM database between 2011–2016 identified 296 sequential test orders (sequencing followed by del/dup if negative), increasing four-fold between 2012–2015. Sequencing identified 70 (24%) pathogenic variants, 24 (8%) variants of uncertain significance (VUS), and 202 (68%) negatives. Of sequencing negative cases, del/dup analysis identified 12 (6%) pathogenic variants and 190 (94%) negatives. For the 70 sequencing positive cases in which del/dup testing was avoided, $874 was saved on average per patient. For the most commonly ordered single genes analyzed by both methods, pathogenic del/dups as a proportion of all positives (not including VUS) were as follows (compared to current GeneReviews estimates in brackets): PTEN 2/2 (100%) [11%], NF1 2/15 (13%) [5%], NSD1 2/4 (50%) [15%], FBN1 0/6 (0%) [unknown], and RB1 1/5 (20%) [12%]. For single genes involved in autosomal dominant or x-linked disorders ordered only once since 2011 using a reflexive strategy (51 total), there were 16 pathogenic variants, none by del/dup analysis.
Conclusion
Over 5 years, meaningful cost savings were achieved through UM-guided sequential testing for inherited pediatric disorders known to be associated with del/dups. The frequency of sequence vs. del/dup alterations varies by gene/disorder, and this data can be collected at the institutional level and compared to literature estimates with the potential to identify novel trends and provide feedback to ordering physicians. While sequencing is useful, separate del/dup analysis in rarer syndromes in which the prevalence of pathogenic del/dups isn’t well described may be best reserved for the research setting.
39. Challenges in Pediatric Pathology Fellowship Recruitment
1University of Michigan, Ann Arbor, Michigan
2Texas Children's Hospital, Houston, Texas
3Children's Medical Center Dallas, Dallas, Texas
4BC Children's and Women's Hospitals, Vancouver, BC
5Children's Mercy Kansas City, Kansas City, Missouri
Background
There is an increased need for more specialized pediatric pathologists due to proliferation of pediatric specific medical knowledge, tests and treatment modalities coupled by the growing pediatric population. Nevertheless, several ACGME-accredited pediatric pathology fellowship openings in North America remain unfilled yearly. The goal of this study is to understand the difficulties in recruitment to pediatric pathology.
Methods
Three separate anonymous surveys targeting Pathology Residents (group 1), Pediatric Pathology Fellows and recent graduates during academic year 2013–2014 (group 2), and Pediatric Pathology Fellowship training programs (group 3) were administered online via Survey Monkey.
Results
The respondents included 175 residents, 29 fellows and 19 Programs. Both residents and fellows reported subspecialty selection early in residency training and primarily based on interest, followed by prospects of employment. 80% of resident respondents had no intent to specialize in pediatric pathology, over 60% said they did not want to be restricted to pediatric diseases or did not wish to deal with pediatric autopsies. Nearly half had discounted pediatric pathology subspecialty training without prior exposure to the specialty. Most residents and fellows learn about subspecialty fellowships from senior residents and faculty members and the choice of the training program was mostly dictated by geographic location. Majority of the fellows had accessed the Society for Pediatric Pathology (SPP) website as residents, but only a few had applied for the resident recruitment award to fund attendance and travel to a SPP meeting. Fellowship training programs interviewed an average of two candidates for each available position and majority of the programs had a fill rate of around 60% averaged over 5 years.
Conclusion
Pathology residents decide on subspecialty training based on their interest; however many are not exposed to pediatric pathology early on in training. Information revealed by the surveys can be used by fellowship programs and SPP to develop strategies to boost recruitment to the specialty of pediatric pathology to meet anticipated increased workforce needs.