
Abstract
The rust layer formed on carbon steel after 1 year exposure to Qinghai salt lake atmosphere was characterised by the following complementary techniques: X-ray diffraction, infrared transmission spectroscopy, scanning electron microscopy and energy dispersive X-ray. The crystalline components of the rust layers consisted primarily of β-FeOOH, γ-FeOOH and iowaite [Mg4Fe(OH)8OCl.4H2O]. δ-FeOOH, ferrihydrite and amorphous rust were also found. Most corrosion products were contained in the inner layer, while foreign particles were mainly distributed in the outer layer. Energy dispersive X-ray and linear scanning results indicated that Cl and Mg elements were rich in the whole rust layer, in which case they will have had an important influence on the corrosion process of carbon steel in the salt lake atmospheric conditions.
Introduction
Atmospheric corrosion is a rather complex interaction in the presence of multiphases between the material and the surrounding atmospheric environment. It is generally ascribed to electrochemical processes occurring on a metallic surface covered by a thin layer of electrolyte under wet/dry conditions. The electrolyte carries various chemicals precipitated from the atmosphere and is of great importance for the corrosion behaviour of metals. Other factors such as temperature, humidity, time of wetness and dust deposition also directly affect the process of atmospheric corrosion of metals due to various atmospheric circumstances. Studies on atmospheric corrosion are traditionally carried out by field exposure tests, which reflect the comprehensive influence of the environment and provide the most reliable information on the process of atmospheric corrosion.
The study of the corrosion products which form on carbon steel is of enormous interest in that it allows us to detect the mechanism of transformation of the rust layer and to evaluate their protective effect.1 – 3 Since the characteristics and structure of the rust layer can greatly affect the subsequent corrosion of the metals, the research on the rust layer has been carried out widely.4 – 12 Generally speaking, the rust is composed of mainly a variety of forms of ferric oxyhydroxides and hydroxides, such as α-FeOOH, β-FeOOH, γ-FeOOH, δ-FeOOH, Fe3O4, etc. These products could co-exist partly as crystalline and partly as amorphous substance, and they also transform to each other in proper conditions. 4 4,5 Stratmann et al.6 have pointed out that the protective effect of the rust layers depended on the chemical composition, compactness, adherence, hygroscopic capacity and morphology. Yamashita et al.5 studied the corrosion products formed on weathering steel exposed to an industrial environment for 26 years and concluded that the main constituent of the rust layer was altered from γ-FeOOH (less than a few years), via amorphous substance (several years), to α-FeOOH (decades). The nanosize α-FeOOH, which formed associated with enriched Cr in the inner layer, was responsible for the protection from atmospheric corrosion. Misawa et al.7 proved the existence of an amorphous ferric oxyhydroxide. Keiser et al.8 showed that the protective rust layer was determined to be composed primarily of δ-FeOOH with a small amount of γ-FeOOH and α-FeOOH and suggested that the presence of δ-FeOOH in the rust layer has a key effect. In a chloride containing environment, β-FeOOH and Fe3O4 were observed in the rust layer. 1 9 1,9,10 Since β-FeOOH has a hollandite structure and Cl− is necessary for the formation of this tunnel structure by locating at the tunnel,9 – 11 it is the representative rust composition in marine environments. Asami and Kikuchi9 investigated the rust on steels exposed to coastal–industrial atmosphere for 17 years, and it was found that the concentration of magnetite was negatively correlated with that of β-FeOOH. Yamashita et al.13 took an alternating wet/dry accelerated corrosion test of steels in the laboratory. It was observed that the content of β-FeOOH in the rust increased with increasing concentration of chlorine ions in the environment, and β-FeOOH was translated from green rust I. Misawa et al.4 and Yamashita et al.13 studied the rust layer formed on weathering steel in marine atmosphere and found that no protective layer formed on the weathering steel because of the erosion of chlorine ions.
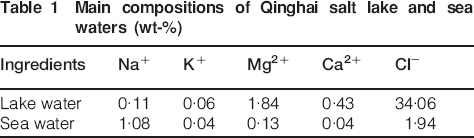
The corrosion rate to be observed on a carbon steel is high in the initial stage of exposure levelling with time.14 This may be due to the fact that the layer of oxide, which is formed on the steel, becomes more compact over time in function of the type of environment and the level of contamination. However, in some specific conditions, especially containing much chloride, it may be unfavourable for the development of protective rust on carbon steel. The Qinghai salt lake, which is located in the northwest of China, possesses an extremely high concentration of chlorine ions. This led to the high deposition rate of sea salt particles in the salt lake region, which facilitated the atmospheric corrosion of carbon steel. The concentration of Cl ions in salt lake water is 34 wt-%, which is 10 times more than that in sea water, and the water possesses a low pH value of 5·0. The compositions of the lake and sea waters are listed in Table 1. As the national western development strategy is gradually implemented, the atmospheric conditions in the Qinghai salt lake region have attracted more and more attention. It is very important to investigate the atmospheric corrosion of carbon steel in this environment in order to reduce costs. In the present study, the rust layers formed on carbon steel after 1 year exposure to Qinghai salt lake atmosphere are characterised, in particular to clarify the role of Mg2+ in the protectiveness of the rust layer and to understand better the mechanism involved.
Main compositions of Qinghai salt lake and sea waters (wt-%)
Experimental
Sample preparation and exposure test
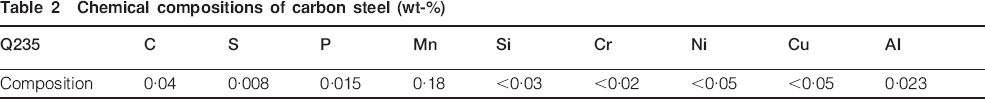
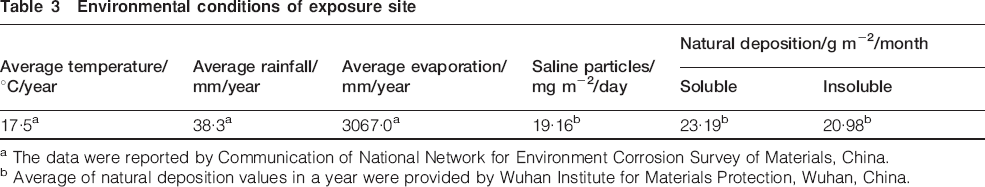
Carbon steel (Q235) was selected as the test material, and its composition is listed in Table 2. The specimens were sectioned into sizes of 100×50×5 mm, polished down to no. 800 grade emery paper, cleaned ultrasonically in acetone, then rinsed with distilled water and dried. The samples were kept in a desiccator for at least 24 h and then weighed using analytic balance (the precision is 0·0001 g). All the samples were exposed to natural atmosphere in the salt lake region. The exposure site was located on the roof of a storage area close to the salt lake. The environmental parameters of the exposure site are listed in Table 3. Specimens were fixed facing south with an angle of 45° to the horizontal. One year later, the samples were taken back for subsequent analysis.
Chemical compositions of carbon steel (wt-%)
Environmental conditions of exposure site
Characterisation
Identification of corrosion products
The rust was scraped off from the metallic substrate by a surgical blade, ground into powder and then stored in a desiccator for a week before the subsequent analysis. Corrosion products were identified by infrared transmission spectroscopy (IRS) and X-ray diffraction (XRD). For IRS analysis, the rust powder was mixed with pure KBr, and the mixture was pressed into a transparent circular flake. A Magna-IR 560 infrared (IR) spectrophotometer was used to determine the IR spectra in the range of 400-4000 cm−1 with an accuracy of 8 cm−1. A step scanning X-ray diffractometer with a Cu target was used for the XRD, with 40 kW intensity, a scanning speed of 2° min−1 and a 2θ range of 15-75°.
Analysis of rust layer
The specimens retrieved from exposure site were stored in the desiccators for analysis. The size of the corroded specimens for SEM observations was 15×15 mm; the elemental composition was determined by energy dispersive X-ray analysis (EDXA). In order to reinforce the rust layer and increase the electrical conductivity, Au was deposited on the rusted surface. The samples for cross-sectional analysis were mounted in epoxy resin at room temperature, mechanically ground down to no. 1500 grade emery paper and polished with a diamond paste of 1·5 μm particle size. Elemental distributions in the rust layer were obtained by linear scanning.
Results and discussion
Rust composition
The IRS and XRD results are plotted in Figure 1 Figs. 1 and 2 respectively. The infrared absorption bands were compared to the reference spectra of various iron oxides 5 5,15 and showed that the rust was mainly composed of β-FeOOH, δ-FeOOH and γ-FeOOH; ferrihydrite and amorphous oxyhydroxide were also detected. The peaks were generally broad and weak, indicating that the corrosion products were poorly crystallised or contained considerable defects. In addition, the absorption band in the vicinity of 1630 cm−1 demonstrated that all corrosion products contained a considerable amount of bound water. 5 12 5,12,16 The XRD patterns contained evidence of the complex compounds iowaite [Mg4Fe(OH)8OCl.xH2O] in addition to the main constituents identified by IRS. Quartz was also found in the rust layer, which may come from dust in the salt lake air. It is worth noting that Mg took part in corrosion reactions in the rust layer; few papers had reported the presence of Mg in the rust. In addition, no a-FeOOH was detected in the XRD and IR spectra of rusts. These must be related to the large amounts of MgCl2 particles in the salt lake atmosphere.
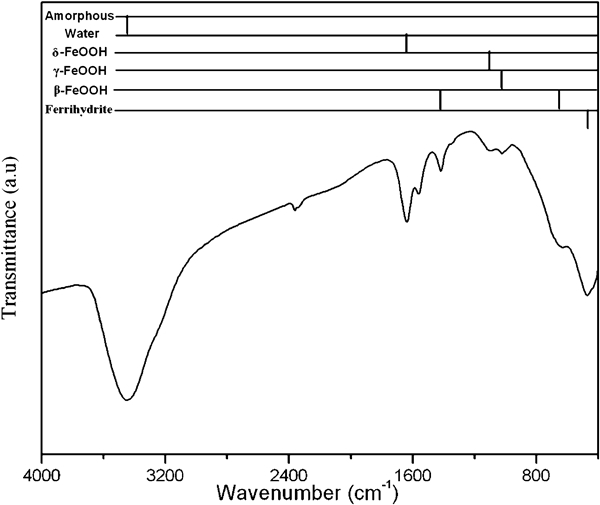
Infrared spectra of rust formed on carbon steel
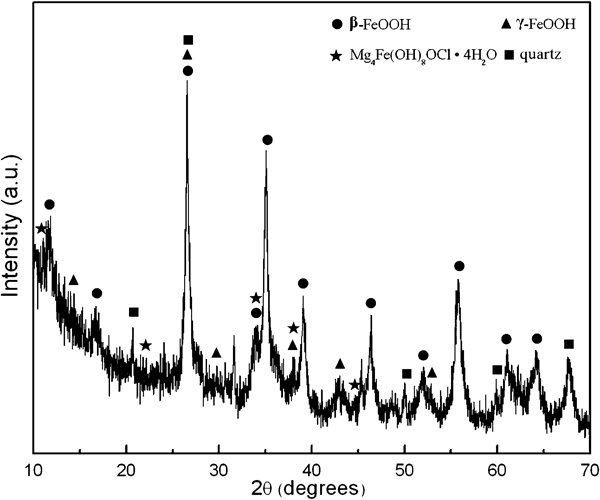
X-ray diffraction patterns of rust formed on carbon steel
The above observations can be explained as follows: according to Kassim et al.,17 the initial oxyhydroxide product in the presence of Cl ions was γ-FeOOH. γ-FeOOH is easily reduced to γ-Fe.OH.OH, 6 6,18 which is an intermediate formed on the surface of γ-FeOOH. In an environment that contains abundant chloride, part of the intermediates may react with Cl− and form green rust I, which can be further transformed to β-FeOOH during the dry stage,10 and part of the intermediates may be oxidised back to γ-FeOOH. Cohen and Hashimoto19 and Keiser et al.20 have studied the electrochemical nature of various ferric oxyhydroxides. They pointed out that α-FeOOH was electrochemically stable, but the other corrosion products, such as γ-FeOOH, β-FeOOH, δ-FeOOH and amorphous ferric oxyhydroxides, were all electrochemically active and could be reduced to intermediates. In the process of dissolution and reduction of corrosion products in the wetting stage, some ferrous ions in the intermediates were replaced by magnesium ions, which could be transformed to iowaite in the reprecipitation process.21 β-FeOOH is usually considered to be the most detrimental to corrosion resistance of steel among ferric oxyhydroxides,22 but iowaite may be actually even more detrimental. The reason is that iowaite [Mg4Fe(OH)8OCl.xH2O] belongs to layered double metal hydroxides, which are known as anion exchangers.23 It has anion ion selective property as well as that of β-FeOOH. However, β-FeOOH is easily reduced and transformed to other phases, while iowaite is less soluble and more stable. Once iowaite was formed, it would work as a reservoir of Cl ions, transfer Cl ions from outer circumstance to the steel substrate incessantly and thus facilitate the corrosion of the steel. On the other hand, it is widely accepted that α-FeOOH is the final stable corrosion product and responsible for the protective ability of the rust. 5 7 5,7,24 Generally, it is present in the rust of carbon steel. However, in this study, it was absent. The reason may be that α-FeOOH is usually transformed from γ-FeOOH in the dissolution–reprecipitation process of the corrosion products, 4 4,7 but in environments containing high Cl− content, the intermediate formed from the reduction of γ-FeOOH would react with Cl− and be further transformed to β-FeOOH. In addition, as analysed above, the formation of iowaite provided a condition of high chloride concentration in the rust layer; thus, the formation of α-FeOOH was suppressed, and it was difficult to detect α-FeOOH in the rust. According to the protective ability index proposed by Kamimura et al.,1 the value of the mass ratio ‘α/γ*’ of crystalline α-FeOOH to the sum of γ-FeOOH, β-FeOOH and Fe3O4 in this work was very low, indicating that the rust had very poor protectiveness.
Analysis of rust layer
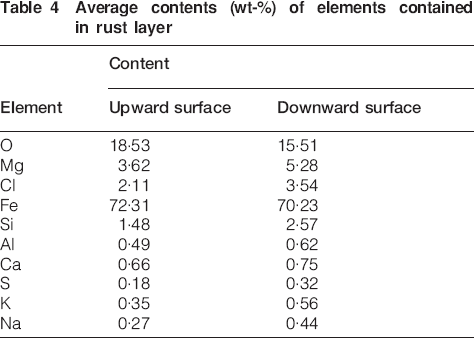
The surface morphology of carbon steel after 1 year exposure to Qinghai salt lake atmosphere was observed by SEM, as shown in Fig. 3. All the exposed samples were completely covered by corrosion products. It is clearly seen that the rust layers formed on both sides were very rough. Compared with that on the upward rust layer, the surface on the downward rust layer contained more cavities and voids, which facilitated the penetration of the impregnant to the substrate, which, in turn, promoted the corrosion process. The average elemental contents in the rust layer were obtained by EDXA, which was carried out on more than five different positions of the rust layer, as summarised in Table 4. The foreign elements O, Mg, Cl, Si, Al, Ca, S, K and Na were detected in both upward and downward rust layers. It indicated that abundant foreign sand particles and contaminations in the air penetrated and were accumulated in the corrosion products, which was not favourable to the formation of a dense protective rust layer on carbon steel. The amount of S was little, while Cl was rich in the rust layer. This illuminated that Cl, rather than S, had an influence on the corrosion of steel in the salt lake region. It is remarkable that a large amount of Mg appeared in the rust on carbon steel, the content of which was the highest among all the foreign cations. This may be due to the fact that Mg was involved in the corrosion reactions, formed insoluble compounds and retained in the rust, which is testified by the above XRD results. The reason why the rest of the foreign cations were not able to participate in the corrosion reactions is analysed as follows: compared with other cations, the ionic radius of Mg2+ is closer to that of Fe2+ and the Mg2+ valence is 2, so only magnesium can substitute for the ferrous ions in the intermediates and form the compound products.21 Thus, other cations may exist in the form of oxide and hydroxide impurities in the rust layer. In addition, foreign elemental contents in the rust of the downward surface were generally higher than that of the upward surface. Usually, the upward surface favoured the deposition of pollutant particles. However, comparing with downward surface, the upward surface suffered a more natural force such as wind and rain, which may lead to less accumulation of foreign particles and lower content of contaminations retained in the upward rust layer.

Scanning electron micrograph of rust layer on carbon steel exposed to salt lake atmosphere
Average contents (wt-%) of elements contained in rust layer
The cross-sectional morphologies of the upward and downward surfaces on rusted carbon steel are shown in Fig. 4. It can be seen that the rust layer formed on the upward surface contained many voids and microcracks and did not generate delamination, which cannot prevent the corrosive species from passing through the rust layer effectively. However, the rust layer formed on the downward surface was comprised of a double layer. The outer rust layer was loose and porous, while the inner rust layer was relatively dense and crack free.

Cross-section morphology of rusted carbon steel
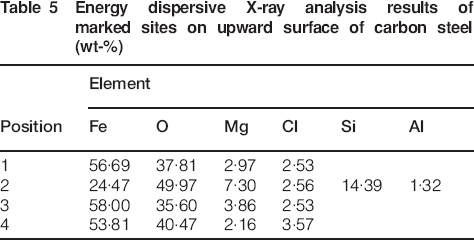
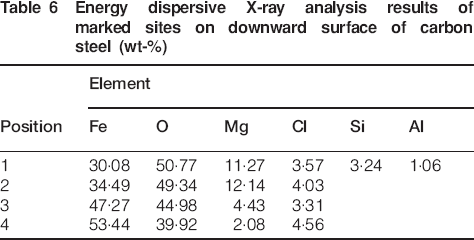
Comparing the rust layer thickness of the upward surface with that of the downward surface, it could be seen that both sides exhibited no apparent difference in corrosion degree. The elemental contents of the marked sites 1-4 on the upward and downward surfaces (shown in Fig. 4) were determined by EDXA semiquantitative analysis, as listed in Table 5 Tables 5 and 6 respectively. It is clearly seen that Fe, O, Mg, Cl, Si and Al were found in both upward and downward rust layers. For both sides, Si and Al only existed in the outer site (position 2 in Fig. 4a and position 1 in Fig. 4b), while Fe, O, Mg and Cl were mainly distributed in the inner sites. This indicated that foreign elements O, Mg and Cl took part in corrosion reactions in the rust layer, while Si and Al existed in the rust layer in the form of dust and impurities, which is in good agreement with the above deduction. The amount of Cl was rich in the whole rust layer, and it had diffused to the interface between the rust layer and the substrate. It is known that the presence of Cl− would facilitate metal dissolution by depassivating the steel surface.25 Therefore, it can be postulated that Cl− had great influence on the corrosion of carbon steel in the salt lake region. In the case of Mg, abundant magnesium accumulated in the corrosion products. These results can be explained by the fact that β-FeOOH and γ-FeOOH form in the vicinity of the steel surface in the initial stage of corrosion and transform into other corrosion products. According to Evans26 and Stratmann and Hoffmann27 and Stratmann and Müller,28 the reduction of the corrosion products would take place at the rust/metal interface. As the intermediate layer grew, Mg was gradually enriched in the rust layer by substituting for some of the ferrous ions in the intermediates. After further reactions, insoluble compound iowaite was formed and retained in the rust. According to the analysis above, due to the fact that the crystallographic structure of iowaite would allow ionic conduction, we can postulate that it may serve as a bridge linking the anodic reaction of Fe dissolution and the cathodic reaction of rust reduction and thus facilitating the corrosion of the substrate.
Energy dispersive X-ray analysis results of marked sites on upward surface of carbon steel (wt-%)
Energy dispersive X-ray analysis results of marked sites on downward surface of carbon steel (wt-%)
The EDXA line profiles of the main elements of the upward and downward surfaces on rusted carbon steel are shown in Fig. 5. Generally, Fe and O were uniformly distributed all over the rust layer. It seemed that a few Si and Al just existed in the outer rust layer, which indicated that they did not influence the corrosion process. Moreover, Cl and Mg were almost distributed in the whole rust layer and enriched near the substrate/rust interface remarkably, which was in good agreement with the detected result by EDX. These facts indicated that both Cl and Mg were involved in the corrosion reactions, formed corrosion products and affected the corrosion process. Based on the theory of ion selectivity, anion (Cl−) can easily enter the rust layer and distribute in the whole rust layer if the rust layer is anion selective. Therefore, it can be postulated that the rust layer has anion ion selective property due to the contribution of iowaite, and it did not effectively hold back corrosive ions. This effect was detrimental for the corrosion of carbon steel, as iron did not form stable corrosion products and protective rust layer in the presence of Cl ions. However, the exact role of Mg cation on the atmospheric corrosion of carbon steel is not fully understood for the time being and needs further observations.
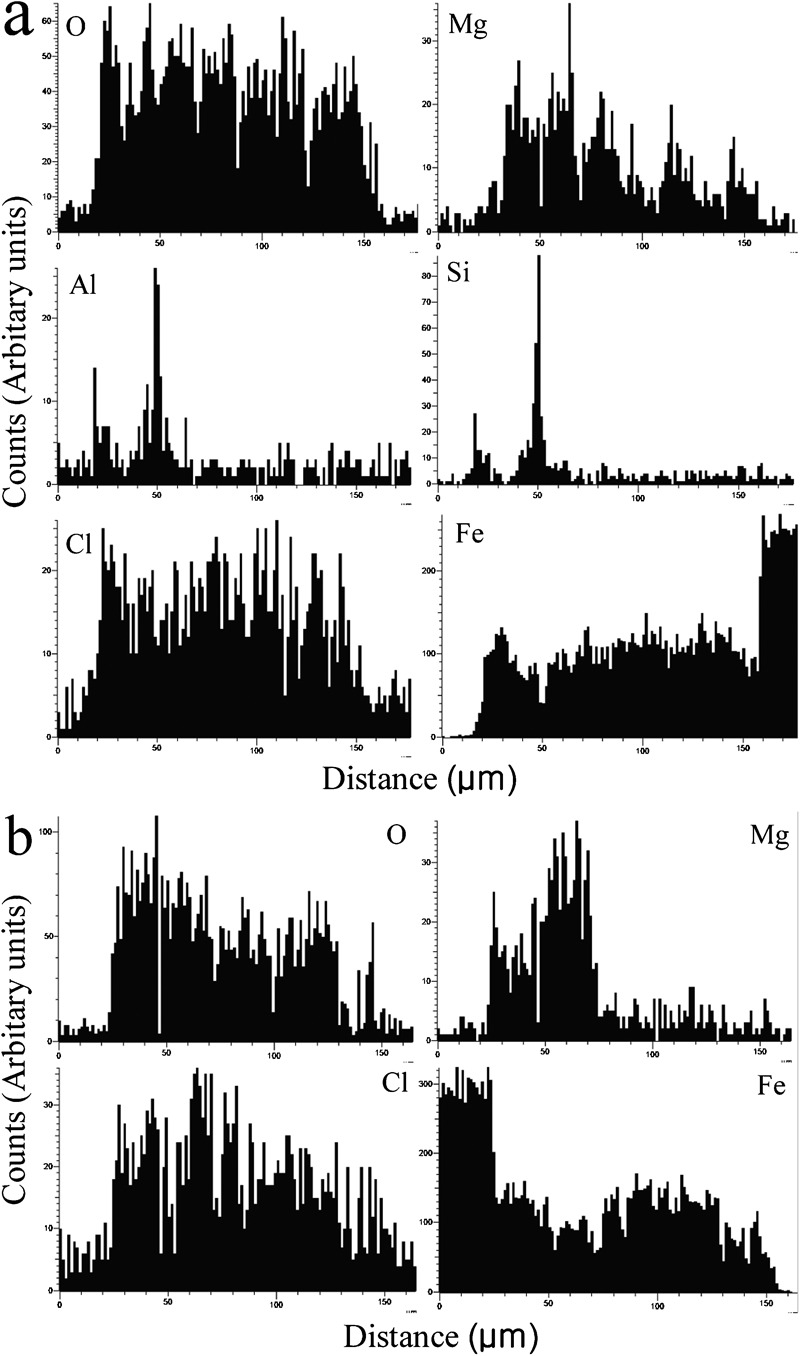
Energy dispersive X-ray analysis line profile of cross-sectional of rusted carbon steel
Conclusions
After 1 year of exposure in the Qinghai salt lake atmosphere, the corrosion degree of the upward surface was almost the same to that of the downward surface for carbon steel. The rust composition was mainly β-FeOOH, γ-FeOOH and iowaite [Mg4Fe(OH)8OCl.4H2O]. The insoluble compound iowaite, which is anion selective, had a great influence on the corrosion behaviour of carbon steel. Cl− was incessantly transferred to the steel substrate through the bridge of iowaite and promoted the corrosion of carbon steel.
Footnotes
Acknowledgements
Financial support for this study was granted by the National Defense Office of Science and Industry (grant no. H102011B002) and the National Key Fund Project (grant no. 51131007).