
Abstract
We recently performed an autopsy on a premature female newborn with rhizomesoacromelic limb shortening of the upper and lower extremities, craniofacial dysmorphism, and chondrodysplasia punctata. A diagnosis of Conradi-Hunermann-Happle syndrome or X-linked dominant chondrodysplasia punctata was made based on elevated cholest-8(9)-ene-3β-ol in serum and tissues. Molecular analysis of EBP, mutations of which are responsible for this malformation syndrome, revealed a monoallelic missense mutation, c.328 G>A (R110Q). We present this case as an illustration of an unusually severe manifestation of this disorder in a female, with additional unusual features including lack of skin manifestations and apparent bilateral symmetry of the skeletal findings.
Keywords
INTRODUCTION
Disorders of postsqualene cholesterol biosynthesis are single-gene metabolic defects that lead to multisystem dysmorphogenesis. The most common of these disorders is the Smith-Lemli-Opitz syndrome (SLOS, OMIM # 270400), which occurs at a frequency of 1 in 60 000 to 70 000 live births in the USA, Canada, and the UK, but at higher frequencies in Central Europe, with an incidence of 1 in 15 000 to 20 000 in Slovakia [1–4]. Smith-Lemli-Opitz syndrome is caused by a deficiency of 3β-hydroxysteroid Δ7-reductase or 7-dehydrocholesterol reductase (EC 1.3.1.21), which catalyzes the reduction of 7-dehydrocholesterol, the final step in the biosynthesis of cholesterol [5–7]. Besides SLOS, 6 other disorders of postsqualene cholesterol biosynthesis have been identified as the etiologies for malformation syndromes (Table 1) [8–13].
Known disorders of post-squalene cholesterol biosynthetic pathway
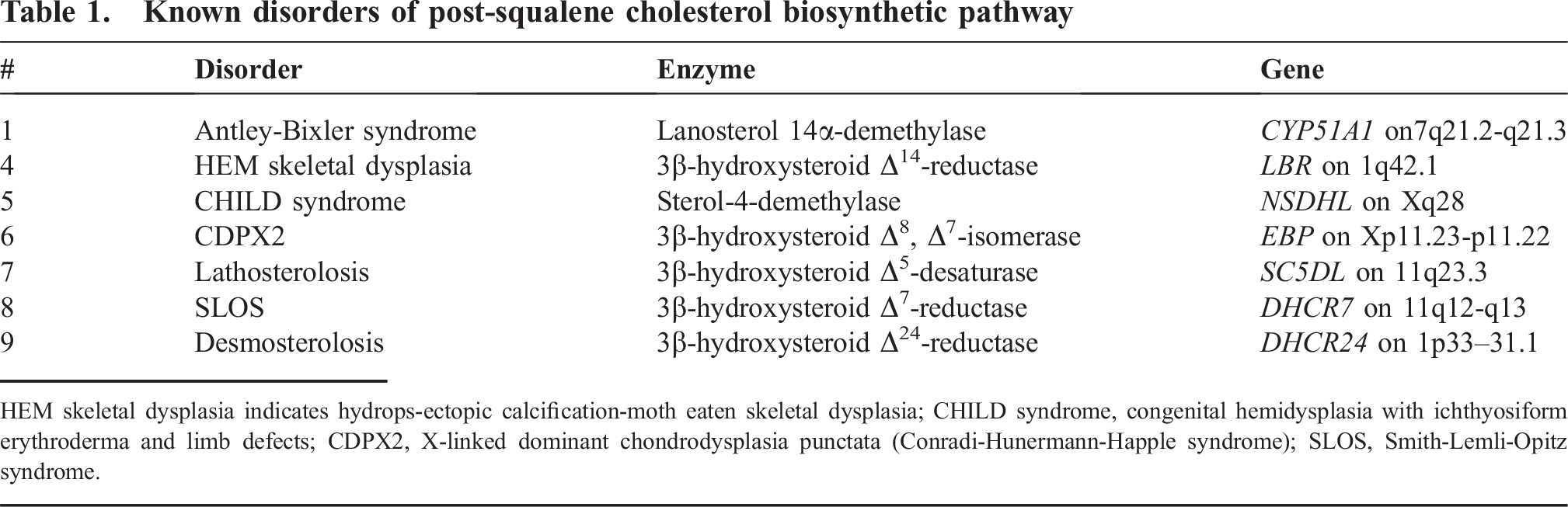
HEM skeletal dysplasia indicates hydrops-ectopic calcification-moth eaten skeletal dysplasia; CHILD syndrome, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; CDPX2, X-linked dominant chondrodysplasia punctata (Conradi-Hunermann-Happle syndrome); SLOS, Smith-Lemli-Opitz syndrome.
Conradi-Hunermann-Happle syndrome (Happle syndrome) or X-linked dominant chondrodysplasia punctata (CDPX2, OMIM # 302960) is an uncommon disorder of postsqualene cholesterol biosynthesis, caused by a deficiency of the enzyme 3β-hydroxysteroid Δ8, Δ7-isomerase (EC 5.3.3.5), which catalyzes the isomerization of cholest-8(9)-ene-3β-ol to lathosterol (Fig. 1) [10, 14–17]. The gene, Emopamil Binding Protein (EBP), which encodes 3β-hydroxysteroid Δ8, Δ7-isomerase, resides on chromosome region Xp11.23-p11.22. A loss-of-function mutation in EBP, leading to the deficiency of 3β-hydroxysteroid Δ8, Δ7-isomerase, produces widespread anomalies and in utero demise of the hemizygous male fetuses. In heterozygous affected females, signs and symptoms of the disorder are milder but variable, presumably because of the random inactivation of either allele. This random lyonization may also account for the bilaterally asymmetric findings in females with CDPX2. The clinical features of CDPX2 include punctate calcifications in epiphyseal cartilages or chondrodysplasia punctata, scoliosis, hemivertebrae, rhizomesomelic limb shortening, patellar dislocation, club feet, ichthyosiform erythroderma, follicular atrophoderma, patchy alopecia with sparse eyebrows and eyelashes, hypoplastic malar eminences, nasal hypoplasia, frontal bossing, abnormal ears with hearing loss, down-slanting palpebral fissures, microphthalmos, cataracts, glaucoma, nystagmus, short neck, tracheal stenosis, Dandy-Walker malformation, mental retardation, hydronephrosis, growth retardation, failure to thrive, fetal hydrops, and polyhydramnios [18]. We recently diagnosed, at autopsy, a severely affected female infant with CDPX2 who had bilaterally symmetric skeletal findings and did not have any skin lesions. Here, we describe the morphologic, biochemical, and genetic findings that led to the diagnosis of CDPX2 in this child.
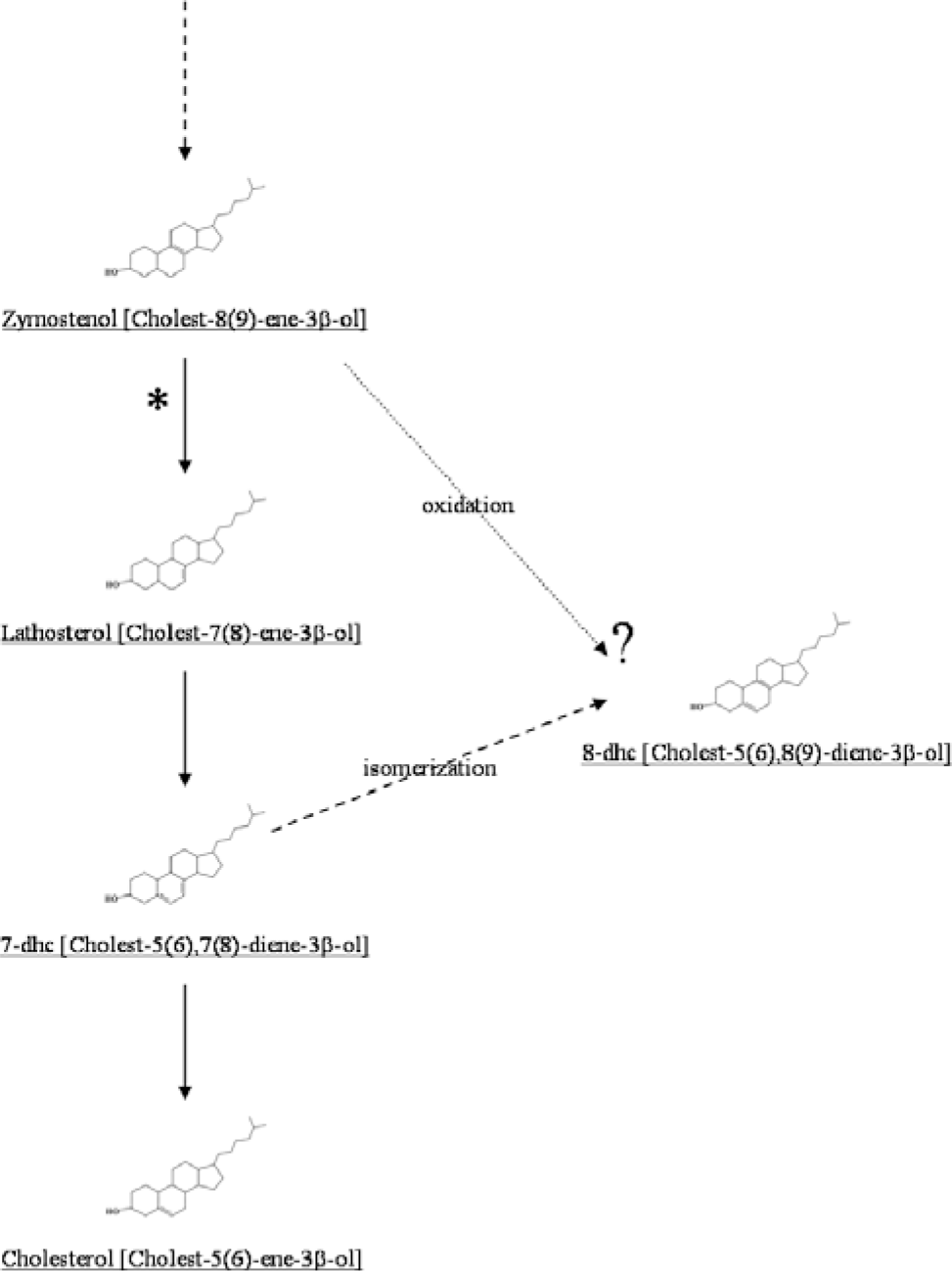
The final steps of cholesterol biosynthesis involving the conversion of cholest-8(9)-ene-3β-ol to cholesterol are shown. The conversion of cholest-8(9)-ene-3β-ol to lathosterol is deficient in patients with X-linked dominant chondrodysplasia punctata. In these patients, there is a diagnostic accumulation of cholest-8(9)-ene-3β-ol in blood and tissues, which can be detected by gas chromatography/mass spectrometry. An accumulation of 8-dehydrocholesterol is also seen in these patients; this may represent oxidation of the excess cholest-8(9)-ene-3β-ol (dotted arrow). In patients with Smith-Lemli-Opitz syndrome, accumulation of 8-dehydrocholesterol is thought to result from isomerization of excess 7-dehydrocholesterol (dashed arrow).
CASE REPORT
The proposita, a 28-week gestation female infant, was born by spontaneous vaginal delivery to a 22-year-old primigravida. An initial fetal sonogram performed at 12 weeks of gestation had confirmed the gestational age estimated by the mother's last menstrual period. There was no history of fever in the mother or any suspected teratogen intake by the mother during the pregnancy. Excessive fundal growth prompted another ultrasound examination at 27 weeks of gestation. At this examination, the ratio of femur length to abdominal circumference was noted to be <0.16, which was interpreted as being highly indicative of the presence of a lethal skeletal dysplasia [19]. The thoracic circumference was <3rd percentile, and there was exaggerated lumbar lordosis and polyhydramnios. At birth, the infant had Apgar scores of 3 and 6 at 1 and 5 minutes, respectively. The child had poor respiratory effort and was intubated for mechanical ventilation. Adequate ventilation proved difficult, which was attributed to suspected pulmonary hypoplasia. The care was redirected and the infant died 2 hours after birth.
At autopsy, the infant weighed 890 g. The child had obvious shortening of all limbs (Fig. 2). The upper and lower extremities were bilaterally symmetrically shortened. In each limb, hypoplasia of all segments contributed to the shortening; that is, the limb shortening was rhizomesoacromelic. In addition, the middle fingers of both hands were also short (brachyphalangy). Other dysmorphic features included brachycephaly, a depressed nasal bridge with nasal hypoplasia, hypertelorism, bilateral talipes equinovarus, and a posteriorly displaced anus. The external genitalia were of a normal female and postmortem cytogenetic analysis confirmed a normal female karyotype of 46,XX. No skin lesions or cataracts were noted. A whole-body roentgenogram showed punctate calcifications involving epiphyseal cartilages of the long bones of all limbs, the short bones of the hands and feet, the ribs, and the pelvis (Fig. 3). Calcifications were also seen in tracheal cartilages and soft tissues of the neck. Internal examination revealed a normal situs. The normally lobated lungs were markedly hypoplastic, with the right lung weighing 4.2 g and the left lung weighing 4.3 g (expected combined weight, 23.7 ± 10.0 g). Histologic examination confirmed the calcifications in tracheal cartilage, epiphyseal cartilages of femur and tibia, and soft tissues of the neck (Fig. 3). Histologic examination of the grossly unremarkable brain revealed scattered ischemic neurons, consistent with terminal hypoxia. All other organs were grossly and histologically unremarkable. In summary, the infant had chondrodysplasia punctata with craniofacial dysmorphism and hypoplastic lungs. The diagnostic considerations included disorders of peroxisomal and cholesterol metabolism.
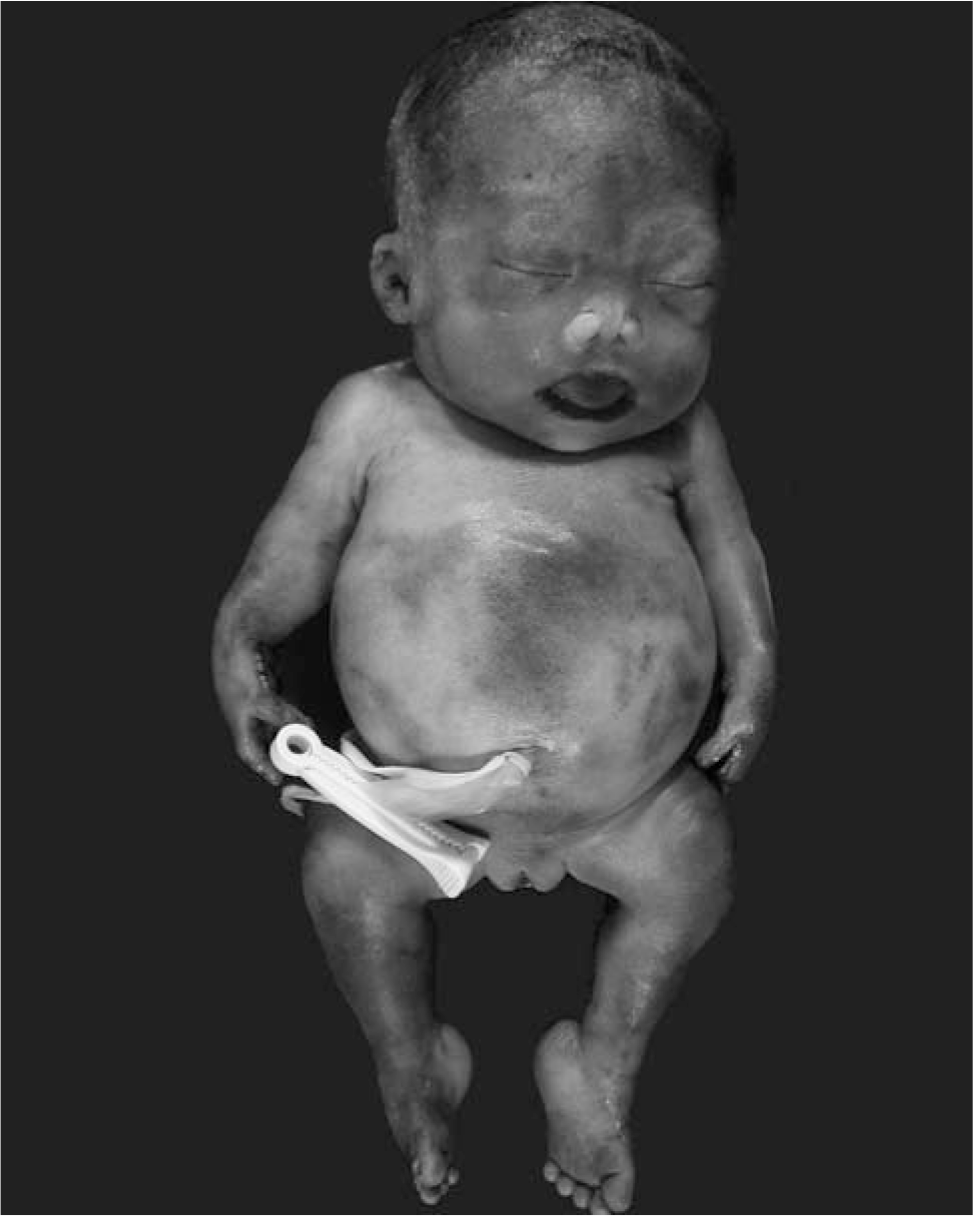
The external malformations seen in our patient with X-linked dominant chondrodysplasia punctata included ocular hypertelorism, hypoplastic nose, short upper and lower extremities, and bilateral talipes equinovarus.
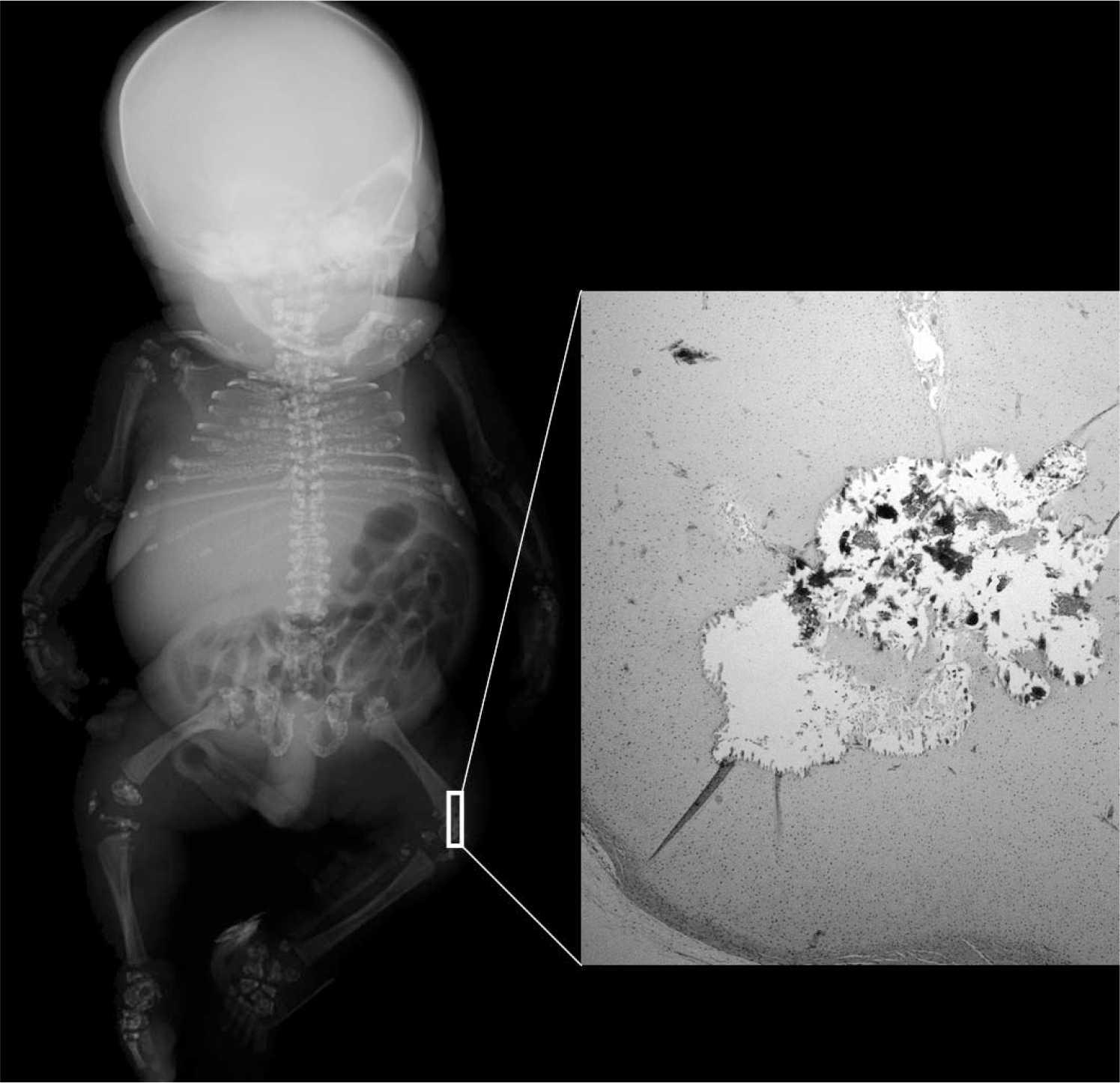
Whole body roentgenogram of the fetus shows stippling of the epiphyses of ribs, pelvis, long bones of upper and lower extremities, and bones of hands and feet. A section from the knee joint shows calcifications (corresponding to the stippling in the X-ray) in the distal femoral epiphysis (hematoxylin and eosin, magnification ×100).
Laboratory workup for peroxisomal disorders
Transmission electron microscopy, performed on glutaraldehyde-fixed liver and kidney samples, revealed structurally and numerically normal peroxisomes. Fibroblast cultures established from a postmortem skin sample were submitted to the Kennedy Krieger Institute in Baltimore, MD, USA for analysis of peroxisomal metabolism. No peroxisomal metabolic defect was identified.
Laboratory workup for cholesterol biosynthetic disorders
Samples of the infant's liver, rib cartilage, brain, and serum were analyzed for the postsqualene cholesterol biosynthetic pathway by gas chromatography/mass spectrometry in the Metabolic Laboratory of the Department of Pathology at Children's Medical Center. All samples revealed abnormal accumulations of the sterol pathway intermediates, cholest-8(9)-ene-3β-ol, lathosterol, and 8-dehydrocholesterol (Figs. 1,4). Quantitation of the sterols in the liver sample showed 95.42% cholesterol, 3.32% cholest-8(9)-ene-3β-ol, 0.94% lathosterol, and 0.32% 8-dehydrocholesterol (Table 2). Accumulation of cholest-8(9)-ene-3β-ol (also called 8(9)-cholestenol or zymostenol) indicates a deficiency of the enzyme 3β-hydroxysteroid Δ8, Δ7-isomerase, which converts cholest-8(9)-ene-3β-ol to lathosterol (Fig. 1), and is diagnostic for CDPX2 [10].
Sterols identified in liver samples from CDPX2 patient and two controls
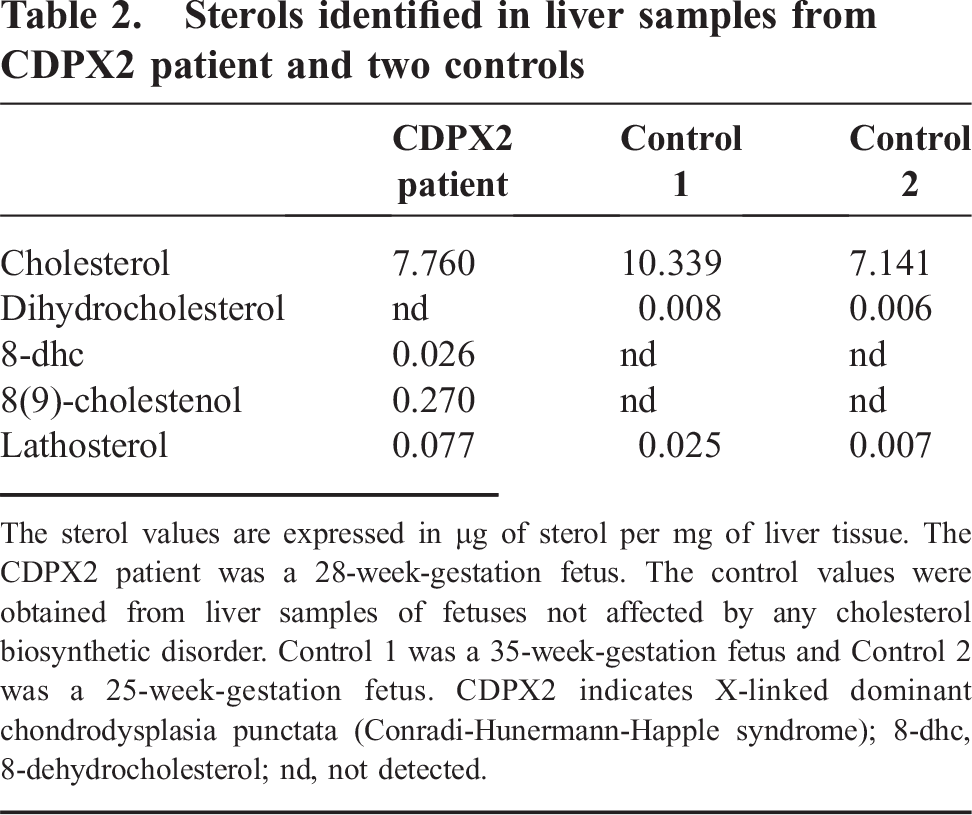
The sterol values are expressed in μg of sterol per mg of liver tissue. The CDPX2 patient was a 28-week-gestation fetus. The control values were obtained from liver samples of fetuses not affected by any cholesterol biosynthetic disorder. Control 1 was a 35-week-gestation fetus and Control 2 was a 25-week-gestation fetus. CDPX2 indicates X-linked dominant chondrodysplasia punctata (Conradi-Hunermann-Happle syndrome); 8-dhc, 8-dehydrocholesterol; nd, not detected.
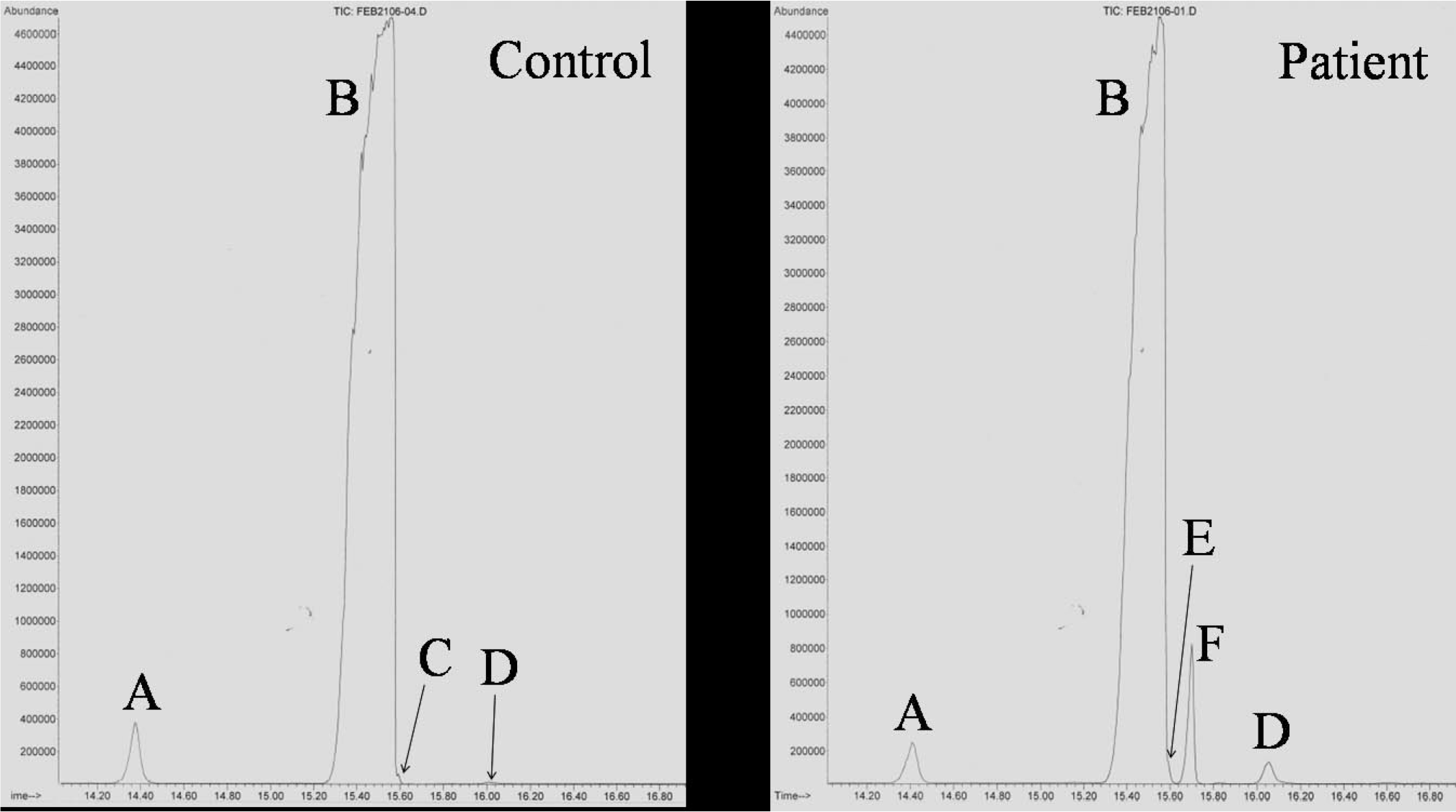
The patient's liver tissue was analyzed for sterols by gas chromatography/mass spectrometry. Gas chromatogram of the control sample on the left shows a large peak of cholesterol (
Molecular analysis of EBP
All 5 exons and intron-exon boundaries of EBP, the gene that encodes 3β-hydroxysteroid Δ8, Δ7-isomerase, were sequenced per Braverman and colleagues [17] at the Advanced Diagnostic Laboratory of the Department of Pathology at Children's Medical Center and the Gene Sequencing Core Facility at the University of Texas Southwestern Medical Center. A mono-allelic missense mutation, c.329G>A, was found in exon 3 (Fig. 5). This mutation would lead to the amino acid change R110Q, wherein arginine is replaced by glutamine at amino acid position 110.
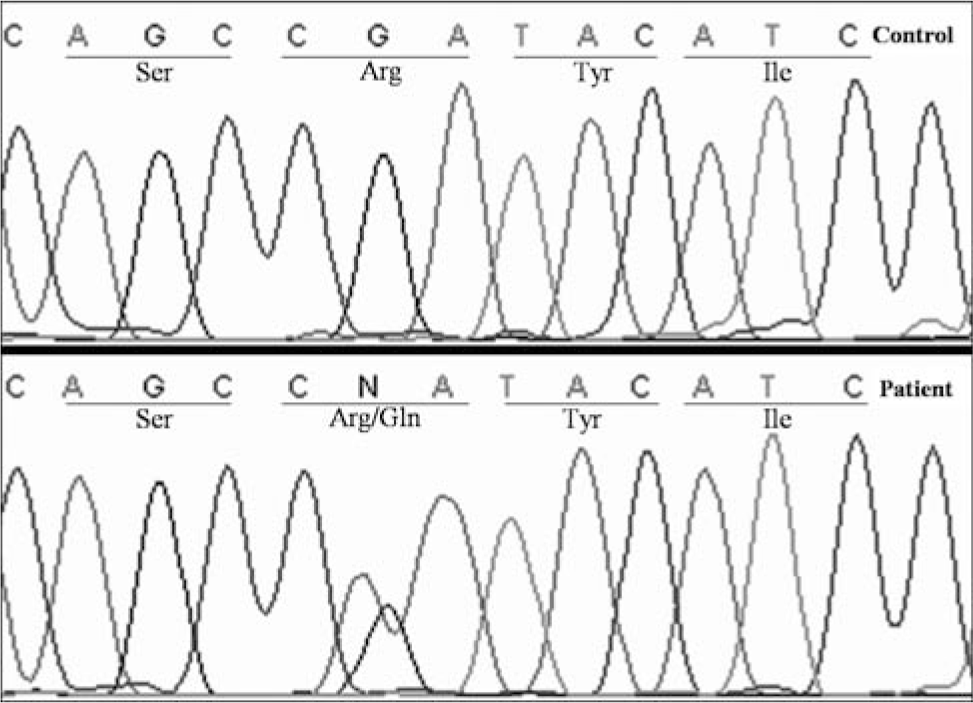
Sequencing of EBP, the gene that encodes 3β-hydroxysteroid Δ8, Δ7-isomerase, revealed a mono-allelic missense mutation, c.329G>A, in exon 3. This mutation would lead to substitution of arginine by glutamine at amino acid position 110 (R110Q).
DISCUSSION
Conradi-Hunermann-Happle syndrome is 1 of 7 postsqualene cholesterol biosynthetic pathway disorders that are currently known to cause multiple malformation syndromes with often devastating consequences for the developing fetus. Conradi-Hunermann-Happle syndrome is caused by a deficiency of 3β-hydroxysteroid Δ8, Δ7-isomerase and is characterized by male fetal lethality and by skeletal, craniofacial, and skin abnormalities that are asymmetrically distributed in heterozygously affected females. The gene that encodes 3β-hydroxysteroid Δ8, Δ7-isomerase, EBP (Emopamil Binding Protein, so named because it binds to the anti-ischemic calcium antagonist, emopamil), resides on chromosome Xp11.23-p11.22, explaining the severe manifestations with early lethality in hemizygous males and the usually milder manifestations in heterozygous females. The random inactivation of X chromosome genes (lyonization) is thought to explain the variable and bilaterally asymmetrical clinical findings in affected females. In the present case of a female infant, the severity of the disorder with early lethality and the symmetrical skeletal features are unusual, probably representing an unusually high percentage of the normal allele being inactivated [20]. Another unusual feature is the absence of obvious skin lesions that characterize CDPX2. It is possible that the characteristic skin lesions and skeletal asymmetry develop later during fetal development and were not manifest in this infant, who was born at 28 weeks of gestational age. This case highlights the importance of considering the diagnosis of CDPX2 in severely affected females with chondrodysplasia punctata.
The lack of skin and hair changes in a patient with chondrodysplasia punctata, limb shortening, and craniofacial dysmorphism including nasal hypoplasia raises the possibility of embryopathy caused by warfarin or other antagonists of vitamin K [21]. It is therefore important to find out whether the mother was being administered any such teratogen during pregnancy; in the present case, the mother was not taking any such drugs. It is also of note that the limb shortening in our patient was not limited to hypoplasia of the proximal segments (rhizomelic), which would be characteristic for rhizomelic chondrodysplasia punctata syndromes that are caused by peroxisomal defects [22]. We did not find any abnormality in peroxisomal metabolism in the present case. The rhizomesomelic limb shortening in CDPX2 was first emphasized by Kelley and colleagues [10]; however, the authors did not state whether or not there was acromelic shortening and brachyphalangy in their reported cases. Conradi-Hunermann-Happle syndrome should also be distinguished from the brachytelephalangic chondrodysplasia punctata, which is apparently inherited in an autosomal fashion [23], and the X-linked recessive brachytelephalangic chondrodysplasia punctata caused by mutation in the arylsulfatase E gene [24].
The missense mutation, c.329G>A (R110Q), identified in the present case, has been described previously by Derry and colleagues [16], who showed that the amino acid arginine at position 110 is conserved through evolution. However, there is a misprint in their article: although the amino acid change R>Q is correctly identified, the base change is incorrectly identified as G>T. Of note, no codon for the amino acid glutamine (Q) has thymine (T) in it. The 2 codons for glutamine are GAA and GAG. In all, 55 mutations of EBP have now been described and include missense, nonsense, deletions, insertions, and splice-site mutations [25–26]. No obvious genotype-phenotype correlation has been noted in the affected females, presumably because of the random nature of lyonization. Conversely, Ikegawa and colleagues [27] have suggested that EBP mutations that produce truncated proteins result in typical CDPX2. On the other hand, the phenotypes resulting from missense mutations, as seen in our case, may result in atypical features. We were unable to perform biochemical or genetic investigations on the mother, who at least clinically appeared to be unaffected. If indeed the mother does not carry the mutated EBP allele in her somatic cells, then either gonadal mosaicism or a new mutation would explain the disease in this case [28]. This is important to know because the probability of the next offspring's being affected would be different in each of these situations.
The pathophysiologic basis for the severe malformations caused by defects in postsqualene cholesterol biosynthesis is being actively investigated. It is known that sterols are precursors for biologically active compounds. For example, cholesterol is a precursor for bile acids, oxysterols, and steroids; 7-dehydrocholesterol is a precursor of vitamin D3; and lanosterol forms meiosis-activating sterols found in germ cells. However, lack of these compounds probably does not explain the widespread malformations. It is now well established that cholesterol plays a role in autocleavage and activation of the hedgehog proteins, which are secreted morphogens involved in body patterning and organogenesis. It is currently believed that a block in the hedgehog signaling pathway accounts for the maldevelopment of the growing fetus in cholesterol biosynthetic disorders [29–30]. It is interesting to note that in the present case, the absolute amount of cholesterol in the liver was not markedly reduced (Table 2) and should have been sufficient to activate the hedgehog signaling pathway; yet the infant suffered from a fatal malformation syndrome. Cholesterol is a ubiquitous component of all biomembranes, and plays a special role in stabilization of lipid rafts, the liquid-ordered membrane microdomains of restricted fluidity. Lipid rafts house biologically important membrane proteins and are involved in cellular functions like signaling, endocytosis, and membrane trafficking [31]. We have previously shown that SLOS lymphoblasts, cultured in cholesterol-free medium, accumulate 7-dehydrocholesterol in their lipid rafts, thereby altering the sterol environment and likely the protein composition of the rafts [32]. We believe that the identification of such membrane lipid raft alterations will shed further light on the pathophysiology of cholesterol biosynthetic disorders.
In summary, we have described morphologic, biochemical, and molecular findings in a new severe case of CDPX2 occurring in a female infant.
Footnotes
ACKNOWLEDGMENTS
The authors thank Dr Richard Kelley for confirming the mass spectrum of cholest-8(9)-ene-3β-ol, and Midori Mitui and Nora Leos for the mutational analysis.