
Abstract

To the Editor,
Smith-Lemli-Opitz syndrome (SLOS) is a complex genetic disorder with distinctive facial features and microcephaly, as well as neurodevelopmental and cognitive difficulties. The etiology of the developmental delay and autistic spectrum features seen in this disorder is still incompletely understood. Past work has shown variations in the severity and specific phenotype of SLOS. Curry et al. [1] distinguished between SLOS I, characterized by longer-term survival, and SLOS II, typified by prenatal mortality. Type II had a higher incidence of postaxial polydactyly, cleft palate, and the occasional distinctive feature of enlarged adrenals. Cherstvoy et al. [2] reported a series in which the age of demise ranged from the newborn period to 17 months of age; the series included severe presentations. In their series, they found that cases with polydactyly often had more notable structural abnormalities of the cerebral cortex. This included disorganization and reduced number of neurons in the hippocampus. They also found that urinary tract abnormalities, such as renal agenesis, were much more common in those who also had polydactyly. Subsequent evidence has shown that the phenotypic spectrum correlates with the severity of the biochemical abnormality and thus the specific genetic mutations in 7-dehydrocholesterol reductase (DHCR7) [3].
In a recent review of congenital brain anomalies in distal cholesterol biosynthesis defects, Hennekam [4] indicated that, in SLOS, the phenotypic spectrum is variable. Most anomalies have been documented by diagnostic imaging, and they range from mere atrophy in various locations to megalencephaly, callosal agenesis, cortical dysplasia, and Dandy-Walker malformation [4].
We recently performed an autopsy on a fetus terminated at 22 weeks for anhydramnios and congenital anomalies (Fig. 1 shows the case alongside a control fetus). Our case had syndactyly of toes 2-3, ambiguous genitalia with an XY karyotype, postaxial polydactyly of the hands, cleft palate (see Fig. 1C), and a facial gestalt consistent with SLOS. In addition to these, our case had an amalgam of the features described by Curry et al. and Cherstvoy et al. [1,2], ie, bilateral renal agenesis and enlarged adrenals. Subsequent genetic testing showed homozygosity for the IVS8-1G>C mutation, which is a known SLOS allele.
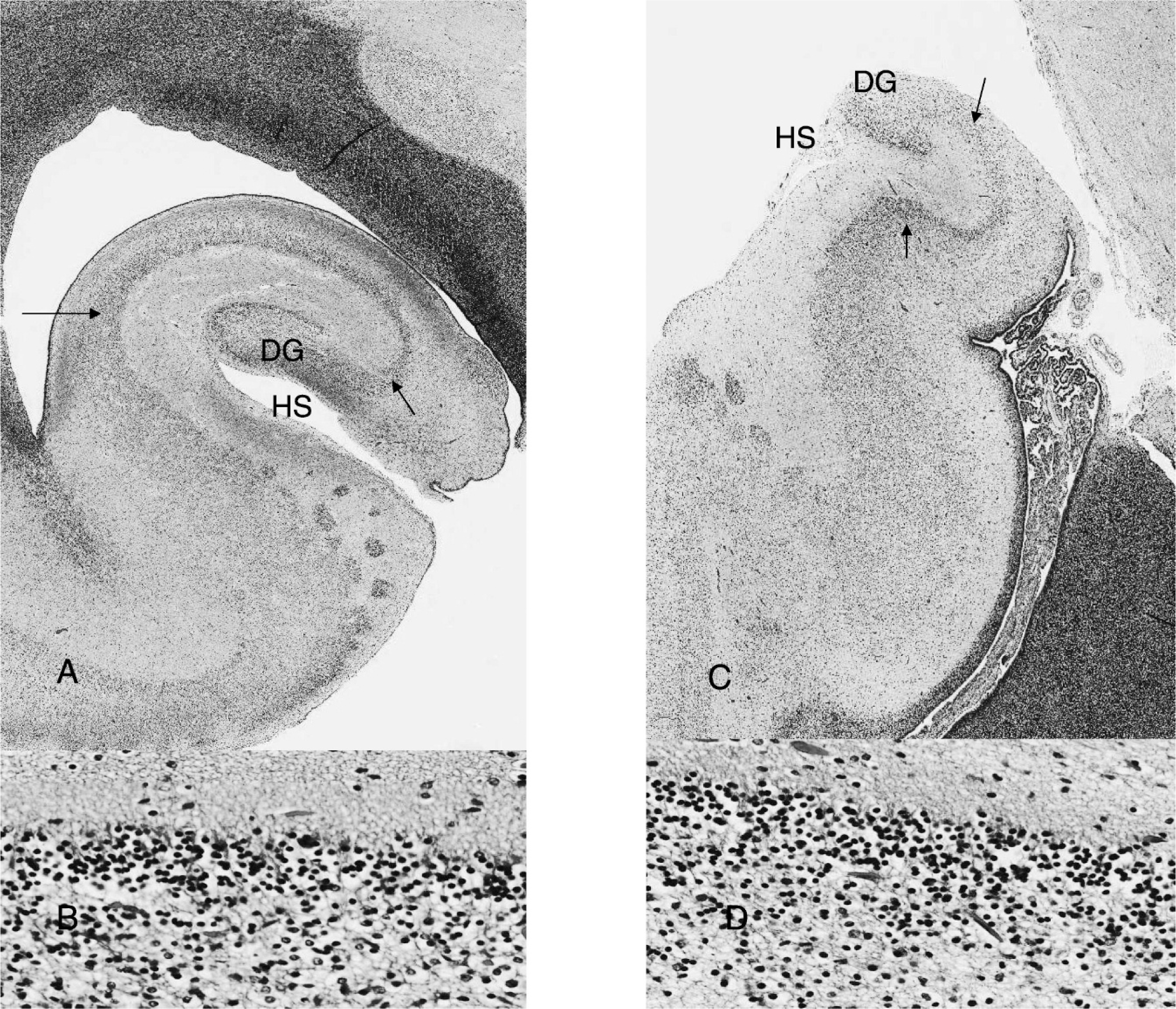
Composite image showing low- and high-power views of hippocampi in a control fetus (
Neuropathological evaluation showed severe hippocampal hypoplasia to an extent that, to our knowledge, has not yet been reported in SLOS (Fig. 1). A control hippocampus from a 23-week-old fetus (Fig. 1A,B), which had very severe renal hypoplasia, was analyzed in parallel using criteria and descriptions reported by Arnold and Trojanowski [5]. The major findings were delayed rotation of the hippocampus, shallow hippocampal fissure, absent internal and external limbs of the dentate gyrus, mild dispersion of immature cells in the pyramidal and polymorph layers of the dentate gyrus, stunted and thicker CA3-CA1 sectors, poorly demarcated limits between the pyramidal layer and the molecular layer and the stratum radiatum, and total absence of emerging pyramidal cells in the deeper layer of the CA1 sector (Fig. 1C,D). This latter finding is intriguing because it suggests another potential anatomical basis for the neurocognitive impairment seen in SLOS. It is possible that this feature is present in only a more severe subset of the condition with a high incidence of early lethality and thus may not be implicated in the neurological and psychiatric symptoms seen in older children. Malrotation of the hippocampus was reported only once following a magnetic resonance imaging study of a 6-year-old boy, suggesting that it may occasionally occur in a less severe subset [6].
One potential mechanism for the effect of the DHCR7 mutation on hippocampal development was provided by Chattopadhyay et al. [7], who showed that impaired cholesterol biosynthesis can adversely impact binding to the major serotonin (5-HT) receptor subtype. This is germane because experimental models have shown that serotonin affects hippocampal development.
Other potential mechanisms discussed in the literature include the effect of abnormal steroid profiles on neurotransmitter and ion-channel function, as well as potential impairment in synaptogenesis and axonogenesis. The overlap between the biologic processes of neural migration and synapse formation invites speculation that more severe forms of SLOS may be associated with overt neuronal hypoplasia/loss, whereas the more subtle forms may display a variety of impaired neural connectivity.
Footnotes
ACKNOWLEDGMENT
The authors wish to thank Lucia Figueredo, Print and Media Specialist, CHEO, for help in compiling the composite image.