
Abstract
Odontogenic myxoma (OM) is a rare, benign, and locally aggressive tumor. It tends to occur in the posterior maxilla and mandible and is often associated with root resorption and perforation of cortex. Histopathologically, there is a proliferation of spindle, bipolar, and stellate cells, with bland nuclei within a myxoid to infrequently fibromyxoid extracellular matrix. Long, thin residual bony trabeculae are often seen floating within the spindle cell proliferation because of the infiltrating nature of this tumor, and these trabeculae impart a “soap bubble” or “tennis-racket” radiologic appearance. No syndromic association of OM has been reported. Although similar histopathologic features are shared with cardiac myxoma and soft tissue myxoma, mutations in the GNAS gene have not been identified in OM to date, and only 2 of 17 OMs showed mutations in the PRKAR1A gene. In this report, we describe a case of OM in a patient with constitutional 1q21 microduplication, a locus that harbors genes encoding certain proteins in the cAMP-dependent protein kinase A (PKA) signaling pathway, including G-protein-coupled receptors and 1 phosphodiesterase interacting protein. Review of the literature describes the key clinical features and molecular pathogenesis of 1q21 microduplication, as well as highlighting the role of PKA signaling pathway in the pathogenesis of myxomas in general.
INTRODUCTION
Odontogenic myxoma (OM) is a rare, benign, slow-growing, and locally aggressive intraosseous tumor. It can be seen in a wide age range but is most commonly seen in the 2nd to 4th decade of life without gender predilection. It occurs anywhere in the maxilla and mandible, with the premolar region of the mandible being the most common location [1–3]. Radiologically, OM is a poorly defined unilocular or multilocular radiolucent lesion with retention of fine, bony septae infiltrated by the tumor, resulting in a “soap-bubble” or “tennis-racket” appearance [4,5]. The lesion infiltrates the adjacent bone and lacks a sclerotic rim. Similar to myxomas of the soft tissue and heart, OM is composed of an unencapsulated proliferation of spindle, bipolar, and stellate cells arranged in a loose myxoid to fibromyxoid background with infiltrative margins. Odontogenic epithelial rests are seen in around 25% of cases [1–3] but are not required for diagnosis. Odontogenic myxomas are predominantly of low cellularity but may have infrequent areas with increased cellularity and collagen intermixed with myxoid matrix. If there is a predominance of fibromyxoid areas, these tumors are referred to as odontogenic fibromyxomas. Perivascular hyalinization may be seen occasionally. Corresponding to radiologic findings, residual fine straight or curvilinear bony septae are embedded within the tumor. The molecular alterations of OM are still under investigation. Genes for 2 major proteins in the cAMP-dependent protein kinase (protein kinase A, PKA) signaling pathway are found to be mutated in soft tissue and cardiac myxomas. Activating mutations of stimulatory Gs alpha subunit gene (GNAS1) have been identified in intramuscular myxomas [6,7], and inactivating mutations of one of the regulatory subunit of PKA (PRKAR1A) have been reported in cardiac myxomas, either sporadically or in the setting of Carney complex [8]. Only 1 case of OM has been reported [9] in a syndromic patient with tuberous sclerosis. There has not been another report of OM in a syndromic or genetically altered background. Here we report a case of OM in a patient with constitutional 1q21 microduplication and discuss the dysmorphism associated with 1q21 microduplication and PKA signaling pathway activation in the tumorigenesis of myxomas in general.
CASE PRESENTATION
A 14-year-old girl presented to the otolaryngology clinic for an expansile lesion involving the left posterior maxilla with tooth displacement. The clinical history included developmental delay, intellectual disability, neuromuscular scoliosis, lung disease secondary to scoliosis, dysmorphic features (severe micrognathia, macroglossia, high arched narrow palate, triangular face), and macular scarring in both eyes. A constitutional chromosomal microarray analysis identified chromosomal 1q21 microduplication, around 13 Mb [arr 1q21.1q22 (144526927156316779) × 3], involving the common recurrent 1q21 microduplication region [10]. Known reference genes are listed in Table 1. In addition, there was a contiguous region of copy neutral absence of heterozygosity (AOH) greater than 10 Mb detected in chromosome 1, adjacent and distal to the identified gain [err 1q22q44 (156188214248346239)] (Fig. 1). Fluorescent in situ hybridization analysis revealed a mosaic duplication involving chromosome 1, with 92 of 110 (84%) cells showing signal duplication.
Reference sequence genes used in chromosomal microarray for band 1q21.1

Genes in the protein kinase A (PKA) signaling pathway are in bold and italic type. CHR indicates chromosome; hg, human genome.
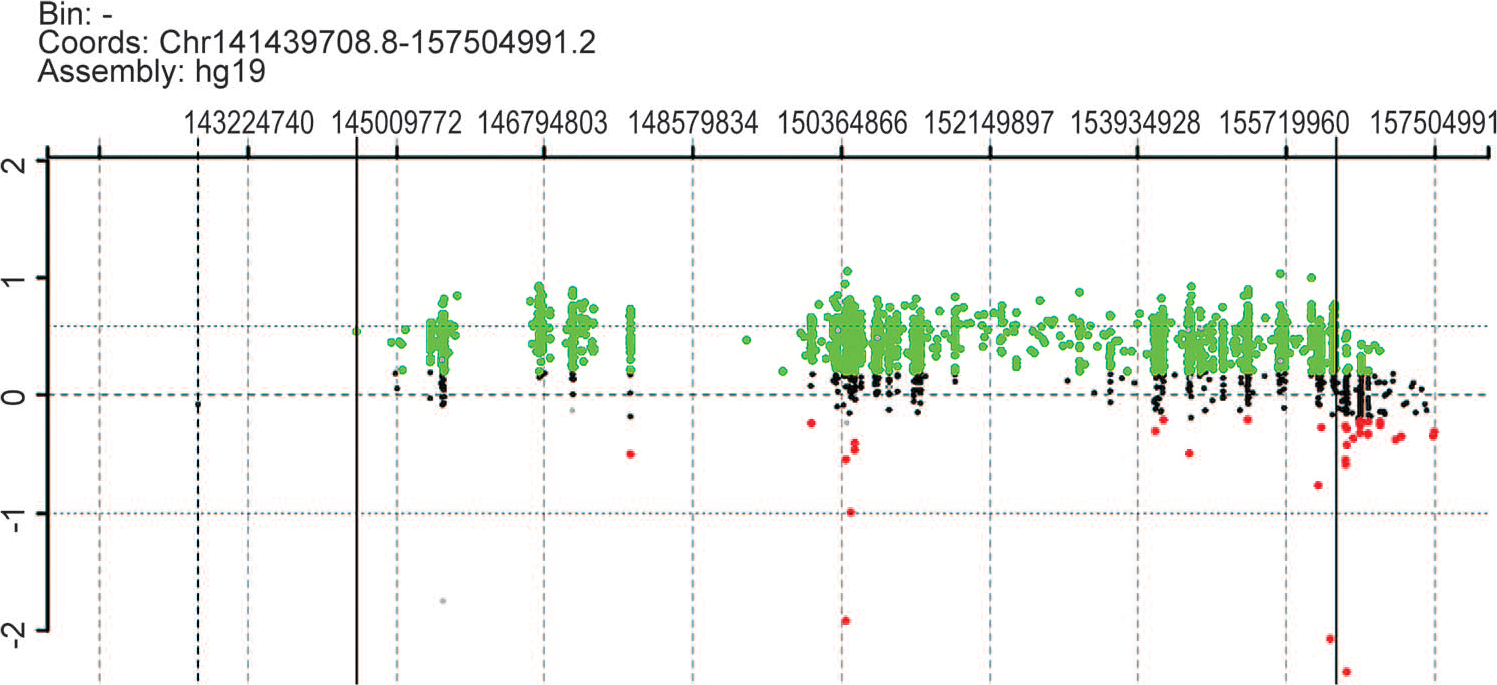
Peripheral blood chromosomal 1: 141439708.8-157504991.2 microarray plot. Probes that had equal copy numbers compared to the control DNA line up on the zero line. Probes that were duplicated moved to approximately 0.58, and the region between 144526927 and 156316779 was duplicated. A color version of this figure is available online.
A maxillofacial computed tomographic (CT) scan showed a large, expansile lytic lesion with internal bony septations and trabeculation involving the posterior left maxilla (Fig. 2), with superior displacement of the left 3rd maxillary molar and medial displacement of the left maxillary premolars and 1st and 2nd molars. The lesion extended posteriorly to involve the left pterygoid plate and superiorly through the inferior and posterior walls of the left maxillary sinus, filling the posterior aspects of the maxillary antrum. The patient underwent an endoscopic nasal sinus and maxillary antrostomy along with an incisional biopsy. Fragments of red-tan to white gelatinous to firm soft tissue were removed in piecemeal fashion with intermixed fine bony spicules, along with the superiorly displaced permanent left maxillary 3rd molar with adherent dental follicle (Fig. 3A). Histopathologically, the biopsy showed spindle and stellate cells embedded in a largely myxoid stroma with only infrequent and focal fibromyxoid areas. The degree of cellularity varied from predominantly sparse to infrequently moderate, with hypercellularity being a focal finding (<5% of all sections examined). The bony septae appeared thin and delicate and occasionally were in parallel arrays. Both reactive woven and lamellar bony spicules were identified. There were only focal portions of tissue with odontogenic epithelial rests embedded in myxoid to fibromyxoid stroma (Fig. 3B–J). Immunostaining of representative tumor, especially the moderately hypercellular area, was negative for S-100 protein, desmin, and epithelial membrane antigen. Electron microscopy was performed on representative tissue from the 1st debulking surgery. The tumor comprised widely spaced, bland spindled to stellate mesenchymal cells embedded in abundant amorphous, somewhat-granular matrix with infrequent to rare collagen fibers (figure not shown). The tumor cells lacked features of nerve sheath cell differentiation, and there were no basal lamina material, myofilaments, z-band material, fibronexus structures, or neurosecretory granules. The tumor cells had the rudimentary features of mesenchymal cells, with typical cell organelles, and lacked dilated rough endoplasmic reticulum, with the granular matrix characteristically seen with fibroblastic cells.
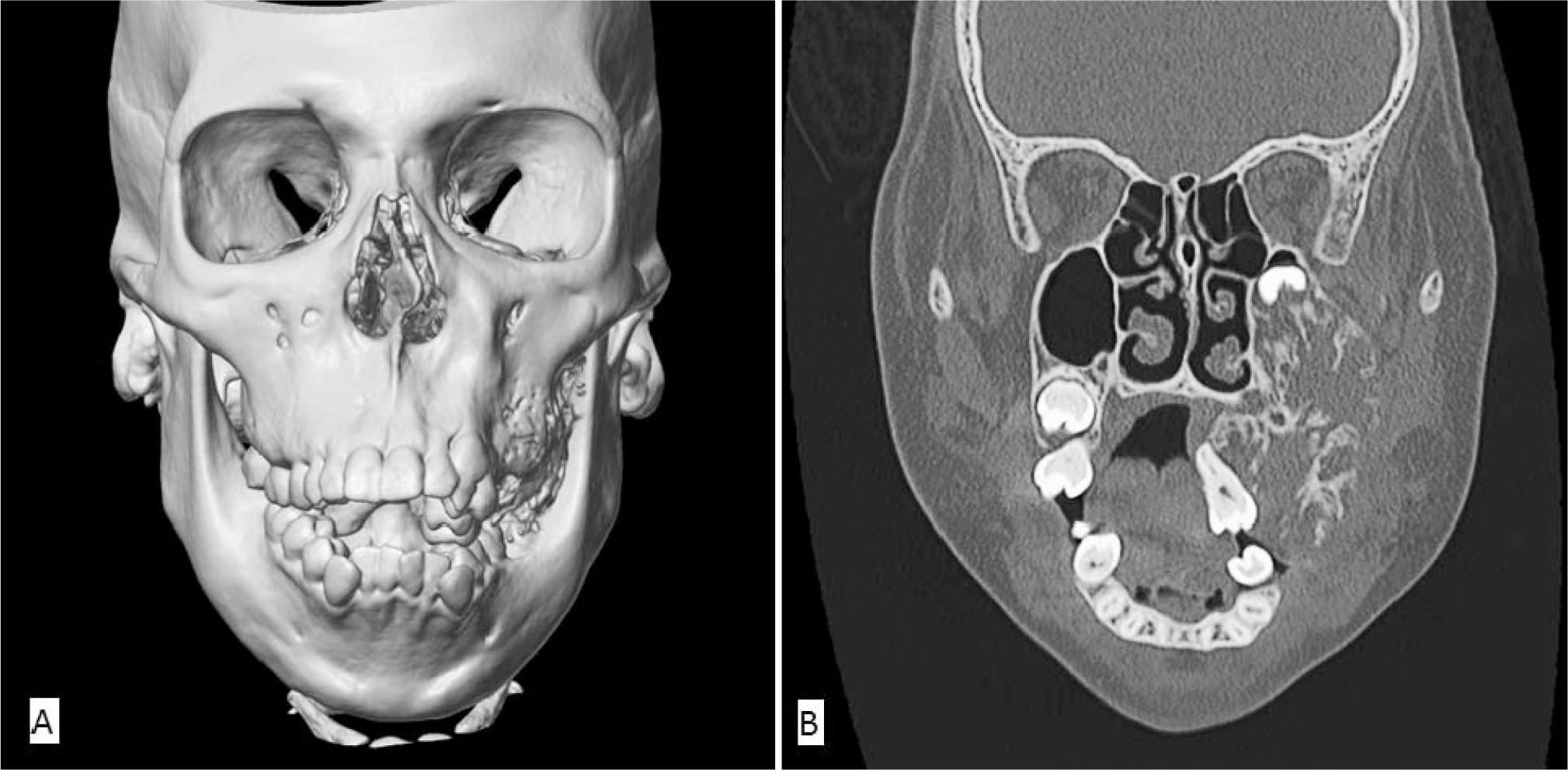
Initial maxillofacial computed tomographic (CT) scan.
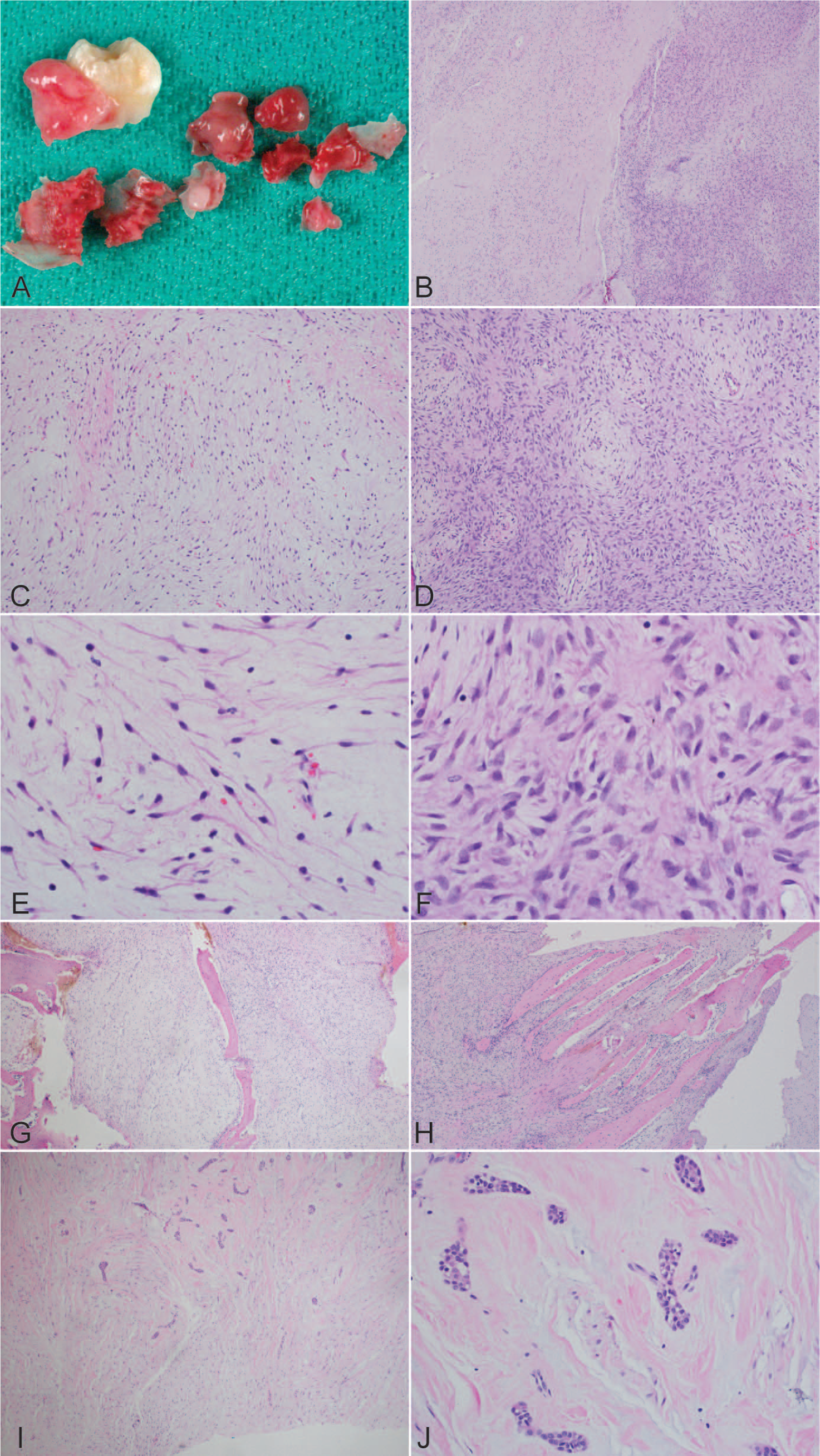
Gross and histologic findings of this odontogenic myxoma.
Three weeks after the diagnostic biopsy, the patient had a left medial maxillectomy, uncinectomy, and partial ethmoidectomy to debulk the tumor because a total resection was difficult to perform given the tumor's large size and the extensive surrounding tissue involvement. A portion of the mass remained, predominantly along the periphery and posterior and medial walls of the maxillary sinus, involving the residual left maxilla and extending along the alveolar ridge and around the roots of the remaining posterior left maxillary teeth (Fig. 4A). A 2nd tumor debulking procedure was performed 11 months later after an interval CT revealed enlargement of the superior aspect of the lesion that extended into the maxillary sinus abutting the maxillary sinus roof(Fig. 4B). Both debulking specimens appeared to be red-tan to white and gelatinous to firm, with intermixed fine bony spicules. There was no interval change in histomorphology, other than absence of odontogenic epithelial rests in either of the debulking specimens.
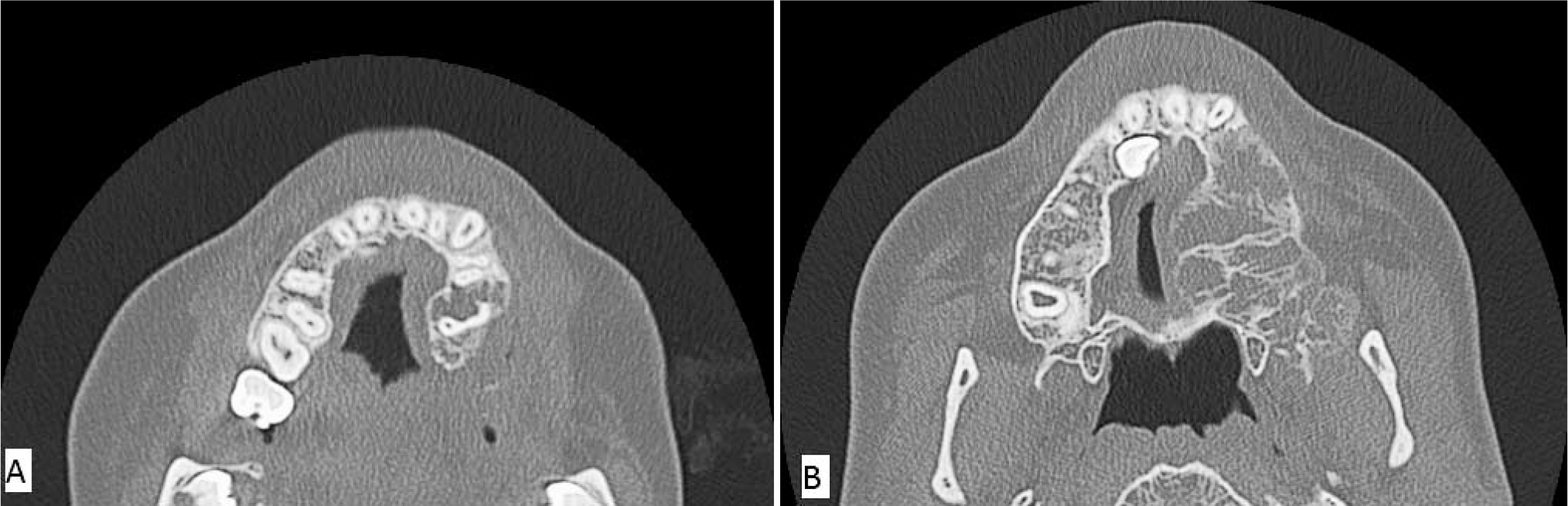
Maxillofacial computed tomographic scan showing continued tumor growth after incomplete initial debulking.
DISCUSSION
Band 1q21.1 includes 5.4 million base pairs. Recurrent chromosomal rearrangement of 1q21 and its phenotypical association were first identified in 2008 [10]. There has been no report of a neoplasm arising in patients with 1q21.1 microduplication in the English literature. General features of 1q21.1 microduplication include abnormal head size along with a spectrum of developmental delay, neuropsychiatric abnormalities, dysmorphic features, and congenital anomalies [10,11]. Additionally, copy number variation at chromosome 1q21.1 has been found in 5 (4 microduplication and 1 microdeletion) of 512 nonsyndromic sporadic tetralogy of Fallot (TOF) cases (P = 0.0002, odds ratio [OR] = 22.3) [12]. Proximal microdeletions of chromosomal band 1q21.1 are also a susceptibility factor for thrombocytopenia-absent radius syndrome, while microdeletions and microduplications of the distal region within 1q21.1 are susceptibility factors for a variety of neurodevelopmental phenotypes [13,14]. Patients with TOF, however, do not have cognitive, social, or neurological disabilities. In fact, family members with the same size microduplication may have very different features. These phenotypic discrepancies are possibly due to different breakpoints, genetic backgrounds, epigenetic imprinting, incomplete penetrance, variable expressivity, and environmental factors. Our patient had certain features associated with 1q21 microduplication, including intellectual disability and facial dysmorphism, but no structural heart defects.
Several candidate genes have been identified to explain certain phenotypes associated with 1q21.1 microduplication. For example, HYD1N gene on 1q21.1 is only active in the brain. Individuals with loss of HYD1N have small head sizes, while individuals with an extra copy of this gene have a large head, suggesting that the HYD1N gene plays a role in determining head size [10,11]. The GJA5 gene is located at 1q21.1 and encodes Connexin 40, a protein that is expressed in the atria of the heart. Duplications of the GJA5 gene are enriched in patients with TOF [15]. A few G-protein coupled receptor (GPR) genes (GPR89A, GPR89B, GPR89C) and 1 phosphodiesterase interacting protein (PDE4DIP) gene are other inhabitants of 1q21.1 (Table 1), and these proteins are key components of the GPR–G protein complex–PKA signaling pathway.
When a GPR is activated by its extracellular ligand, it transmits the signal to an attached intracellular heterotrimeric G protein complex and activates GNAS of the stimulated G protein complex. The activated GNAS binds to and activates adenylyl cyclase, which, in turn, catalyzes the conversion of adenosine triphosphate into cAMP—thereby increasing cAMP levels. cAMP activates PKA by binding to and detaching the regulatory self-inhibitory subunits of PKA, subsequently exposing the 2 catalytic subunits. The exposed catalytic subunits phosphorylate downstream molecules. There are 4 isoforms of the regulatory subunits (PKARIA, RIB, RIIA, RIIB) [16]. After being hydrolyzed by cAMP-specific phosphodiesterases (PDE), specifically, PDE4, PDE7, and PDE8, the cytosol cAMP decreases in amount, and there is subsequent quiescence of the PKA signaling pathway [17].
GNAS1 mutations are found in 30–60% of intramuscular myxomas, with mutational hotspots R201H and R201C being equally affected, the former being the most common mutation in McCune-Albright syndrome, which has significant clinical overlap with Mazabraud syndrome, a syndromic condition that tends to have multiple intramuscular myxomas [18]. The presence of GNAS1 mutations has not been extensively tested in Mazabraud syndrome, possibly as a result of its rarity. Mutation of R201H causes a constitutively activated form of GNAS, which leads to high adenyl cyclase activity and cAMP levels and, subsequently, higher PKA activity [6,19].
Mutations of GNAS1 have not been detected in atrial myxomas [7]; instead, these tumors often exhibit inactivating mutations in PRKAR1A [8,20]. The presence of PRKAR1A mutations in atrial myxomas appears to be more prevalent in patients with Carney complex than in sporadic cases [8]. In addition to atrial myxomas, up to one third of patients with Carney complex also have cutaneous (eyelids, nipples, external auditory canal, ear lobes, and perineum) myxomas [21]. All of these myxomas share similar histomorphology to intramuscular myxomas. To date, 2 Carney complex predisposition loci have been identified (CNC1 and CNC2), with the PRKAR1A gene located in the CNC1 locus. Up to 70% of patients with Carney complex harbor PRKAR1A mutations [22]. More than 120 disease-causing PRKAR1A mutations have been reported in Carney complex. The majority of mutations are subject to nonsense-mediated messenger RNA decay, leading to decreased amounts of protein and, as a result, activated PKA signaling [23].
In summary, either activating mutations of GNAS1 or inactivating mutations of PRKAR1A are found in myxomas at several sites, and all of these mutations result in activation of the PKA signaling pathway. Because of its morphologic similarity to soft tissue myxomas, mutational analysis of GNAS1 has been attempted in odontogenic myxomas, but no GNAS1 mutations were identified in 7 cases tested [24]. In a similar study [25], 17 cases of odontogenic myxoma were subjected to PRKAR1A immunohistochemistry (IHC) and coding region sequencing [25]. Two cases demonstrated mutations in PRKAR1A, 1 missense mutation and 1 frameshift mutation. Both cases showed decreased PRKAR1A protein expression in the tumor tissue by IHC. In addition, 7 of the 15 patients without mutations showed minimal to absent PRKAR1A in tumor cells compared to surrounding nontumor tissue, suggesting the presence of additional mutations that can escape the detection by coding region sequencing, epigenetic changes, intronic alterations that may alter RNA splicing or protein expression, as well as secondary protein modifications. Alternatively, mutations in other genes that ultimately result in the activation of PKA pathway may be the underlying molecular changes in cases of myxomas without detectable mutations in PKA1R1A or GNAS. For example, gene amplification or activating mutations of GPR may lead to the same myxoma phenotype.
CONCLUSION
Even though we cannot exclude the possibility of an unrelated coincidence, the presence of multiple genes in the PKA pathway in chromosome 1q21.1 raises the possibility of genetic alterations playing a role in the development of an odontogenic myxoma in this patient with constitutional 1q21.1 microduplication. It is anticipated that further investigations looking for mutations in all key players of the PKA signaling pathway may yield insight into the molecular mechanisms of myxoma development.
Footnotes
ACKNOWLEDGMENT
We thank Ms Karen Prince for her help with graphic work.